Prader-Willi syndrom
Clemens Gödel er frilanser for det medisinske teamet
Mer om -ekspertene Alt -innhold kontrolleres av medisinske journalister.Prader-Willi syndrom (PWS) er et resultat av en medfødt defekt i arvematerialet. Berørte spedbarn er korte, mentalt underutviklede og muskuløse. I tidlig barndom utvikler de en umettelig sult som fører til uttalt fedme. De resulterende sykdommene skal behandles gjennom terapi fra ulike medisinske spesialiteter. Les mer om symptomene, diagnosen og behandlingen av Prader-Willi syndrom her!
ICD -koder for denne sykdommen: ICD -koder er internasjonalt anerkjente koder for medisinske diagnoser. De finnes for eksempel i legebrev eller på attester om arbeidsuførhet. Q87
Prader-Willi syndrom: beskrivelse
Prader-Willi-syndromet (feilaktig også Willi-Prader-syndromet) ble beskrevet for første gang i 1956 av barneleger Andrea Prader, Alexis Labhart og Heinrich Willi. Omtrent en av 20000 nyfødte lider av Prader-Willi syndrom. Årsaken er en genetisk utløst dysfunksjon av hypothalamus, et viktig byttesenter i hjernen. Manifestasjonen av Prader-Willi syndrom kan være veldig annerledes og kompleks.
Prader-Willi syndrom: symptomer
Berørte fostre blir lagt merke til allerede før fødselen. De beveger seg merkbart lite i livmoren. Pulsen er lavere enn normalt. På fødselstidspunktet er det mer sannsynlig at fostre med Prader-Willi syndrom er i unormale stillinger i mors kropp. Babyer trenger mye støtte under og etter fødselen.
Umiddelbart etter fødselen skiller de berørte nyfødte seg ut på grunn av en stillesittende livsstil, (muskel) svakhet og lav fødselsvekt. Den typiske skrikingen etter fødselen kan også være fraværende eller bare svak. På grunn av den uttalte svakheten og de resulterende suge- og svelgeforstyrrelsene, synes babyene det er vanskelig å drikke.
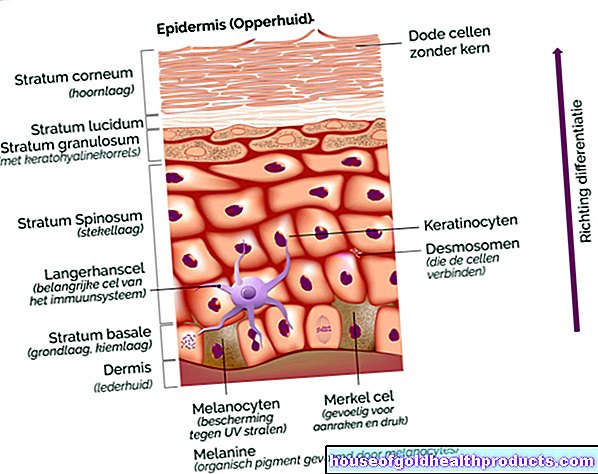
Babyer med Prader-Willi syndrom viser ofte visse ytre egenskaper. Det smale ansiktet er preget av mandelformede øyne og en liten munn med en tynn overleppe. Skallen er ofte lang (dolidochocephaly), og hendene og føttene er små. Ryggraden kan bøyes til en S-form (skoliose). Beinsubstansen i hele kroppen viser skader og defekter (osteoporose / osteopeni) i røntgenbildet. Pigmentering av hud, hår og netthinne kan reduseres. Synsforstyrrelser og mysestillinger (strabismus) i øynene er også mulig. Pungen er liten og ofte tom (testamenter uten nedstigning). Samlet sett er utviklingen av de syke barna forsinket.
På slutten av det første leveåret forbedres muskelsvakheten noe. Imidlertid er det alltid minst en svak svakhet. Normale aktiviteter kan raskt bli slitsomme og slitsomme for pasienter med Prader-Willi syndrom. Likevel liker barn like mye typiske barndomsaktiviteter. Veksten er vesentlig redusert i småbarnsalderen.
Uhemmet matinntak
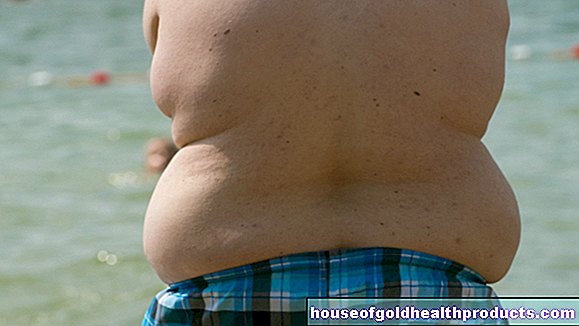
Etter hvert som sykdommen utvikler seg, spiser berørte barn mer og mer (hyperfagi) - uten å føle seg mette. Mat blir hamstret og, selv i alvorlige tilfeller, stjålet. Det er ekstremt vanskelig for barn å kontrollere spisevanene sine. Derfor utvikler de en tydelig fedme (fedme). De går opp i vekt spesielt raskt fordi det er et markant avvik mellom det høye kaloriinntaket og det lave energiforbruket.
Overvekt fører til typiske sekundære sykdommer: hjertet og lungene lider av fedme. En fjerdedel av alle som ble rammet utviklet diabetes mellitus ved 20 -årsalderen. Søvnforstyrrelser, venøse sykdommer (tromboflebitt) og vannretensjon er også blant mange mulige konsekvenser. I løpet av sykdommen kan det oppstå søvnforstyrrelser. I tillegg til gjentatte pustestopp, er forstyrrelser i dag-natt-rytmen eller dyp søvn mulig. Berørte barn synes det er vanskelig å lære.
Utviklingen av puberteten er forstyrret
Den typiske vekstspurt i puberteten er liten. Berørte barn vokser vanligvis ikke høyere enn 140 til 160 centimeter. Selv om puberteten kan begynne for tidlig (for tidlig adrenarke), slutter utviklingen av puberteten i mange tilfeller aldri. De berørte forblir vanligvis sterile. Hos gutter forblir penis og spesielt testiklene små. Det er ingen pigmentering og rynker på pungen. Det kan hende at stemmebrudd ikke forekommer. Hos jenter forblir kjønnsleppene og klitoris underutviklet. Den første menstruasjonsblødningen forekommer ikke i det hele tatt, for tidlig eller sent, noen ganger bare mellom 30 og 40 år.
Mental og mental utvikling
Både mental og psykomotorisk utvikling er forstyrret i Prader-Willi syndrom. Milepæler i barns utvikling nås ofte senere enn hos friske jevnaldrende. Språklig og motorisk utvikling tar noen ganger dobbelt så lang tid som hos friske jevnaldrende.
Den gjennomsnittlige intelligenskvoten (IQ) er mellom 60 og 70 og dermed godt under normen. Den fysiske svakheten kan gjøre det vanskelig å snakke. Taleutviklingen er forsinket og taleforståelsen svekkes, avhengig av lav IQ. Rundt 40 prosent av de syke er på randen av intellektuell funksjonshemming. Uavhengig av IQ dukker det også opp læringsforstyrrelser som aritmetiske vansker. Skolens prestasjoner er stort sett under gjennomsnittet.
Både utvikling av følelser og atferd kan være merkbar. Berørte mennesker blir noen ganger beskrevet som sta og raske. Psykiatriske abnormiteter kan allerede oppstå i tidlig barndom: former for såkalt opposisjonsatferd samt stiv og besittende oppførsel er beskrevet. Rutinemessige prosesser kan være vanskelige. Noen ganger må prosesser gjentas tvangsmessig. Autistiske trekk forekommer hos rundt 25 prosent av pasientene. Attention Deficit Disorder (ADD) er også vanlig.
Symptomene øker vanligvis med alder og overvekt. Hos eldre voksne kan imidlertid symptomene på Prader-Willi syndrom lett gå tilbake. Rundt ti prosent lider av psykose. I tillegg er epilepsier og former for "søvnavhengighet" (narkolepsi) forbundet med Prader-Willi syndrom.
Prader-Willi syndrom: årsaker og risikofaktorer
Årsaken til Prader-Willi syndrom antas å være en dysfunksjon av en del av mellomhjernen kjent som hypothalamus. Blant annet resulterer dette i en mangel på det viktige veksthormonet. Lidelsen skyldes i omtrent tre fjerdedeler av tilfellene mangelen på et gen-segment på kromosom 15 (15q11-q13). I Prader-Willi syndrom synes en defekt i farens kromosomkopi å være av spesiell betydning (70 til 75 prosent) ) slik at det bare er en kopi av genet. En annen mulighet er at begge genene i det doble settet med kromosomer bare kommer fra moren (uniparental disomi, 25 til 30 prosent). En såkalt "pregningsdefekt" er mindre vanlig (en prosent). Begrepet "imprinting" beskriver det faktum at gener leses avhengig av opprinnelse (på mors eller fars side).
I de fleste tilfeller er lidelsen ikke arvelig. Det utvikler seg vanligvis bare under utvikling av kimceller eller etter befruktning. På den annen side er det imidlertid mulig at eksisterende genmutasjoner (for det meste såkalte balanserte translokasjoner) også forårsaker Prader-Willi syndrom. I disse tilfellene øker risikoen for arv.
Prader-Willi syndrom: Undersøkelser og diagnose
Alle nyfødte med vedvarende og uforklarlig svakhet bør testes for PWS. Selv nyfødtlegen (neonatologen) eller barnelegen (barnelegen) vil mistenke Prader-Willis syndrom etter fødselen på grunn av barnets oppførsel. Ingen såkalte prediktive tester utføres uten bevis på tilstedeværelsen av dette syndromet. Imidlertid er det fullt mulig å diagnostisere Prader-Willi syndrom allerede før fødselen.
Fysiske og tekniske undersøkelser
I de fleste tilfeller gir den fysiske undersøkelsen allerede en høy mistanke om Prader-Willi syndrom (Holms Criteria 1993, 2001). Hovedkarakteristikken ved Prader-Willi syndrom er uttalt svakhet, noe som er spesielt tydelig når man drikker. Utseendet gir også ledetråder. Normalt er detekterbare reflekser bare svakt uttalt.
En målbar mangel på veksthormon i blodet er også diagnostisk nyttig. Kjønnshormoner (østrogen, testosteron, FSH, LH) er også stort sett redusert hos de som rammes. Dette er ledsaget av en underutvikling av kjønnsorganene. Binyrbarkens funksjon er forstyrret i mange tilfeller. Dannelsen av kjønnshormoner (androgener) kan også begynne for tidlig (tidlig adrenarke).
Undersøkelsen av hjernebølgene (elektroencefalogram, EEG) kan også være mistenkelig.
Genetisk undersøkelse
Genetiske tester utføres for å bekrefte mistanken om Prader-Willi syndrom. I det første trinnet blir metyleringen av det avgjørende punktet på kromosom 15 (15q11.2-q13, "SNRPN locus") undersøkt. Enzymer kan binde såkalte metylgrupper til DNA og derved modifisere det. I over 99 prosent av tilfellene gir denne undersøkelsen diagnosen. Ellers utføres en annen vanlig metode for å påvise kromosomendringer, fluorescens in situ hybridisering (FISH).
Lignende sykdommer er Martin Bell syndrom eller Angelmann syndrom. Lidelsen i Martin Bell syndrom er på X -kromosomet (skjørt X -syndrom). Ved Angelmann syndrom og Prader -Willi syndrom er den samme posisjonen på kromosom 15 slettet i de fleste tilfeller - men i Angelmann syndrom bare den på mors kromosom.
Prader-Willi syndrom: Behandling
Det finnes ingen kur mot Prader-Willi syndrom. Imidlertid kan symptomene lindres ved hjelp av strengt veiledet, støttende terapi. Hovedkomponentene i behandlingen er matkontroll, hormonbehandling og behandling av atferdsproblemer.
næring
Tilstrekkelig ernæring og tilstrekkelig vekst må sikres, spesielt hvis muskelsvakheten er alvorlig. Prober eller spesielle kunstige brystvorter kan brukes for å lette inntaket av mat. I tillegg bør det utarbeides en fôringsplan med et veldokumentert kaloriinntak og vektkontroll.
Men når barna blir eldre, utvikler de en lidelse for overspising. Deretter må en plan med en streng kaloribegrensning følges. En fettbegrensning er ikke alltid rettet mot, siden fett er viktig for hornutvikling. Spiseforstyrrelsen kan kreve streng håndtering av mat. Spiseforstyrrelsen ved Prader-Willi syndrom har en særlig alvorlig effekt på sykdomsforløpet, ettersom de som rammes beveger seg lite og det dermed er stor avvik mellom kaloriinntak og behov. Det er avgjørende å involvere foreldrene intensivt og tilby det syke barnet en solid struktur.
Samtidig må det sikres at nok vitaminer og mineraler tilføres.I Prader-Willi-syndromet oppstår ofte forstyrrelser i beinmetabolismen, som kan forebygges med vitamin D og kalsiuminntak. Skjelettutvikling, spesielt ryggraden, bør overvåkes regelmessig.
Aktiviteten og motoriske utviklingen av det syke barnet blir jevnlig undersøkt og om nødvendig støttet terapeutisk med fysioterapi eller lignende metoder.
Veksthormon
Fra toårsalderen kan veksthormonet HGH også gis som et stoff. Denne behandlingen bør stoppes når vekstplatene i beinet er lukket (røntgenkontroll). Hormonbehandling bør alltid overvåkes nøye. Hormonadministrasjonen har en positiv effekt på kroppens utvikling, men bivirkninger er fotødem, forverring av krumningen i ryggraden (skoliose) eller økt trykk i skallen (pseudotumor cerebri). Pusteforstyrrelser kan oppstå i starten av behandlingen. Det er derfor viktig å overvåke søvn i starten av behandlingen. Overvåking av HGH-hormonbehandling inkluderer også regelmessige målinger av skjoldbruskkjertelenivåer og blodnivåer av vekstfaktoren IGF-1.
Ved pubertetsforstyrrelser kan kjønnshormoner administreres som depotinjeksjoner, hormonplaster eller gel. Dette kan forbedre atferdsproblemene. Østrogener hjelper også med å bygge bein, men de har også en rekke bivirkninger.
Psykologisk støtte
Det berørte barnet bør få hjelp til å støtte oppførsel og utvikling av milepæler i utviklingen. Spesielt sosiale ferdigheter må trenes i Prader-Willi syndrom. Interaksjon med jevnaldrende og omsorgspersoner bør også oppmuntres. En-til-en-veiledning kan være nødvendig på skolen. Om nødvendig må bo- og arbeidsmiljøet tilpasses. Psykiatriske abnormiteter kan kreve medikamentell behandling, for eksempel med en serotoninantagonist. Målet med den intensive støtten er best mulig uavhengighet.
Kirurgiske forsyninger
Enhver leppe og ganespalte som allerede kan eksistere ved fødselen, kan behandles av kirurger på et tidlig stadium. Feiljusteringer av øynene, spesielt skjelingstillinger, kan også behandles kirurgisk for å forhindre synsforstyrrelser. Først av alt kan midlertidig dekning av det friske øyet hjelpe.
Underutviklingen av kjønnsorganene kan kreve kirurgi for å flytte testikkelen fra nedre del av magen til pungen. Beta-hCG (humant koriongonadotropin) kan forstørre pungen og la testikkelen synke.
Skjelettendringer i Prader-Willi syndrom kan også behandles kirurgisk. En S-posisjon i ryggraden (skoliose) krever kirurgisk korreksjon i alvorlige tilfeller, men blir vanligvis behandlet uten operasjon (for eksempel med et korsett.
Prader-Willi syndrom: sykdomsforløp og prognose
Den tidligste diagnosen "Prader-Willi syndrom" kan ha en positiv effekt på den langsiktige prognosen. En positiv innflytelse på (spise) atferd og eventuell administrering av veksthormoner kan forbedre livskvaliteten til det berørte barnet.
Spiseatferd, vekt og vekst bør kontrolleres med jevne mellomrom etter diagnosen. Utvikling, funksjon, atferd samt psykiatriske abnormiteter kontrolleres. Tett observasjon og omsorg av en spesialist som kan koordinere og sikre tverrfaglig omsorg er svært viktig.
Syke har økt risiko for infeksjoner flere ganger. Spesiell forsiktighet må utvises under operasjoner: Etter anestesi tar vekkefasen ofte lengre tid og det er økt risiko for pusteforstyrrelser.
Det største problemet er imidlertid økende fedme. I løpet av livet dominerer de resulterende komplikasjonene. Dødeligheten øker også på grunn av komplikasjonene. Den økte dødeligheten ved Prader-Willi syndrom skyldes derfor først og fremst hjerte-, vaskulære eller lungesykdommer.
Arverisikoen er lav. I de fleste tilfeller er den genetiske endringen spontan og ikke arvet. Risikoen for foreldre for å få et andre barn med Prader-Willi syndrom er lav. De berørte forblir ofte barnløse.
Bare i sjeldne tilfeller er Prader-Willi syndrom forårsaket av endringer i den genetiske informasjonen i foreldres kromosomsett, som er arvelige og også fører til større sannsynlighet for arv. Hvis et barn har Prader-Willi syndrom, anbefaler vi at foreldre som ønsker å få barn, søker råd fra menneskelige genetikere.
Tags.: boktips symptomer uoppfylt ønske om å få barn










.jpg)