Marfans syndrom
Mareike Müller er frilansskribent i medisinsk avdeling og assisterende lege for nevrokirurgi i Düsseldorf. Hun studerte humanmedisin i Magdeburg og fikk mye praktisk medisinsk erfaring under utenlandsoppholdet på fire forskjellige kontinenter.
Mer om -ekspertene Alt -innhold kontrolleres av medisinske journalister.Marfans syndrom (MFS) er en genetisk sykdom i bindevevet. Pasientene har forskjellige symptomer i ulik grad: lange fingre og smale, lange lemmer eller blodåreskader. Det er ingen kur mot Marfans syndrom. Regelmessige kontroller kan forhindre komplikasjoner. Les alt om Marfan syndrom her!
ICD -koder for denne sykdommen: ICD -koder er internasjonalt anerkjente koder for medisinske diagnoser. De finnes for eksempel i legebrev eller på attester om arbeidsuførhet. Q87
Marfans syndrom: beskrivelse
Marfans syndrom er en genetisk sykdom som enten overføres fra foreldre til barn eller utvikler seg spontant. En sykdom som utvikler seg spontant er også kjent som en sporadisk sykdom. Dette gjelder omtrent 25 til 30 prosent av pasientene med Marfans syndrom. Totalt sett er en til fem av 10.000 mennesker i befolkningen rammet av Marfan syndrom. Det er ingen forskjell mellom kjønnene.
Marfan syndrom: symptomer
Tegnene på Marfan syndrom er veldig forskjellige og forskjellig uttalt hos individuelle pasienter. Selv i samme familie kan symptomene på Marfan syndrom variere sterkt blant de syke familiemedlemmene. Ulike organsystemer påvirkes av sykdommen. Det vanligste er endringer
- Sirkulasjonssystem
- skjelett
- øye
Marfan syndrom: Kardiovaskulært system
Pasienter med Marfan syndrom har økt risiko for plutselig død. Årsaken til dette er en hyppig forekommende rift i veggen i hovedpulsåren (aortadisseksjon). Som et resultat av dannelsen av et gap i aortaveggen, transporteres blodet ikke lenger inn i de mindre blodårene, men siver heller inn i hullene. Risikoen for aortadisseksjon er økt hos pasienter med Marfan syndrom fordi aorta, som har svekkede vegger, gradvis utvides (progressiv aortadilatasjon).
I tillegg lider pasientene ofte av hjerteklaffskader som aorta- og mitraloppstøt. Disse kan føre til hjertearytmier. Videre risikerer de betennelse i hjertet (endokarditt) og hjertesvikt.
Marfan syndrom: skjelett
Skjelettendringer er ofte det første tegnet på Marfans syndrom. Pasientene skiller seg ut for sin høye vekst og veldig smale, lange ekstremiteter. Edderkoppfingre (arachnodactyly) er et velkjent symptom. Edderkoppfingrene kalles slike fordi de er ekstremt lange og smale.
I tillegg har mange pasienter misdannelser i brystet, for eksempel et kylling- eller traktbryst. Som ytterligere endringer i skjelettet lider de ofte av skoliose, bøyning og vridning av ryggraden. I tillegg har noen pasienter underutviklede ansiktsbein, for eksempel kinnben eller overkjeven.
Hele disse skjelettendringene er også kjent som marfanoid habitus.
Marfan syndrom: øye
Endringene i øyet forårsaket av Marfans syndrom påvirker hovedsakelig linsen. Det blir ofte forskjøvet (lins ektopi). Dette truer pasienten med blindhet. En annen risikofaktor for blindhet er nærsynthet. Det er forårsaket av et for langt øyeeple. Denne endringen kan også føre til netthinneløsning.
Marfans syndrom: symptomer som påvirker andre organer
I tillegg til de nevnte organsystemene, kan Marfans syndrom også skade andre strukturer. Dette inkluderer blant annet lungene. De som er rammet har økt risiko for å utvikle pneumothorax. Leger forstår at dette betyr løsrivelse av lungehinnen fra pleura og luftinntrengning i dette gapet. Denne tilstanden kan være livstruende ettersom lungene kollapser i det berørte området.
Strekkmerker blir ofte sett på huden til pasienter med Marfan syndrom som et tegn på svakt bindevev.
I løpet av livet kan en såkalt duraectasia utvikle seg. Dette er en forlengelse av hjernehinnene, vanligvis på nivået av korsryggen. Det er ofte asymptomatisk. I noen tilfeller kan det forårsake smerte når dural sac presser på de spennende spinalnervene.
Marfan syndrom: årsaker og risikofaktorer
Marfans syndrom er en autosomal dominant, arvelig sykdom. Dette betyr at det er en endring (mutasjon) i et gen i vår genetiske sammensetning som utløser sykdommen. Autosomaldominant beskriver at denne genetiske informasjonen ligger på et ikke-kjønnsspesifikt genkompleks (autosomalt) og alltid vises (dominerende).
Når en pasient med Marfan syndrom får et barn, kan han eller hun arve enten det syke eller det sunne genet. Fordi hver person har et dobbelt sett med genetisk sminke. Dette betyr at overføringssannsynligheten er 50 prosent. Et barn av en pasient med Marfan syndrom har 50 prosent sjanse for sykdommen.
Marfan syndrom: skadet bindevev
Mutasjonen som forårsaker Marfan syndrom er på den lange armen til kromosom 15 (15q21). Det påvirker det såkalte FBN1-genet. Dette genet er ansvarlig for dannelsen av et bindevevsprotein, fibrillin-1. Fibrillin-1 er viktig for stabiliteten i bindevevet. Hvis dannelsen er begrenset av mutasjonen, mister bindevevet stabilitet.
Marfan syndrom: forskjellige former
Alvorlighetsgraden av Marfans syndrom varierer. Leger snakker da om variabel ekspressivitet. Dette betyr at symptomene på pasientene også er forskjellige i en familie. Til tross for den samme mutasjonen kan en pasient knapt ha symptomer, mens et søsken viser hele bildet av Marfan syndrom.
Marfans syndrom: undersøkelser og diagnose
Diagnosen Marfan syndrom stilles ofte av en barnelege. Totalt sett spiller ulike spesialister en rolle i diagnostisering, behandling og råd. I tillegg til barnelegen inkluderer dette menneskelige genetikere, kardiologer, ortopeder og øyeleger. Før diagnosen endelig blir stilt, vil legen din først spørre deg i detalj om din medisinske historie (anamnese). Han vil stille deg følgende mulige spørsmål, blant andre:
- Har et familiemedlem Marfan syndrom?
- Føler du av og til et racing -hjerte?
- Har du alltid vært høyere enn andre da du var barn?
- Er du nærsynt?
Marfan syndrom fysisk eksamen
Legen din vil da gjøre en fysisk undersøkelse. Da ser han først på skjelettet nøye. Han tar hensyn til lengden på de enkelte beinene, brystets form og ansiktsformen. Så lytter han til hjerte og lunger. Hjertearytmier eller strømningslyder kan bli lagt merke til over hovedpulsåren.
For å stille diagnosen Marfan syndrom ble de såkalte Gent-kriteriene utviklet. Den viser forskjellige symptomer på sykdommen i forskjellige former. Når et visst antall kriterier er oppfylt, kan diagnosen stilles.
En genetisk test av Marfan syndrom er også mulig. Den genetiske sammensetningen analyseres og det søkes etter mutasjonen som er ansvarlig for sykdommen. Hvis det er tilfeller av Marfan syndrom i familien, kan en passende diagnose stilles før fødselen.
Marfan syndrom: Lignende kliniske bilder
Andre genetiske sykdommer som kan føre til lignende symptomer må skilles fra Marfan syndrom. Disse inkluderer blant annet
- Ehlers-Danlos syndrom
- Loeys-Dietz syndrom
- Sphrintzen-Goldberg syndrom
- MASS syndrom
Marfan syndrom: behandling
Siden Marfans syndrom er en genetisk sykdom, kan selve årsaken, mutasjonen, ikke behandles. Målet med terapien er en regelmessig kontroll av pasienten av forskjellige spesialister for å forhindre komplikasjoner. Viktigst er hjerteovervåking viktig for å forhindre plutselig død av aortadisseksjon. Dette kan oppnås ved å motvirke aorta -utvidelse ved å administrere betablokkere og begrense fysisk aktivitet. Aorta -utvidelsen kan overvåkes ved årlige ultralydundersøkelser, og aortaroten kan korrigeres i god tid før disseksjonen.
Ytterligere operasjoner som kan være nødvendige i tilfelle Marfan syndrom
- Skoliose korreksjon
- Brystkorreksjon
- Fjernelse av linse
Marfan syndrom: sykdomsforløp og prognose
Sannsynligheten for at mutasjonen overføres fra en forelder til et barn er 50 prosent. Par med en partner med Marfan syndrom som planlegger å få barn, bør søke råd fra en menneskelig genetiker.
I dag er forventet levetid og livskvalitet nesten ubegrenset for pasienter med Marfan syndrom. Selv om forventet levealder var klart begrenset tidligere, har den blitt økt med 30 år de siste 30 årene. Pasienter har imidlertid fortsatt økt risiko for aortadisseksjon, noe som kan føre til plutselig død. Aortadisseksjon er oftest sett rundt 30 år. Regelmessige kontroller av behandlende spesialister kan redusere risikoen for aortadisseksjon ved Marfans syndrom.
Tags.: Sykdommer sykehus laboratorieverdier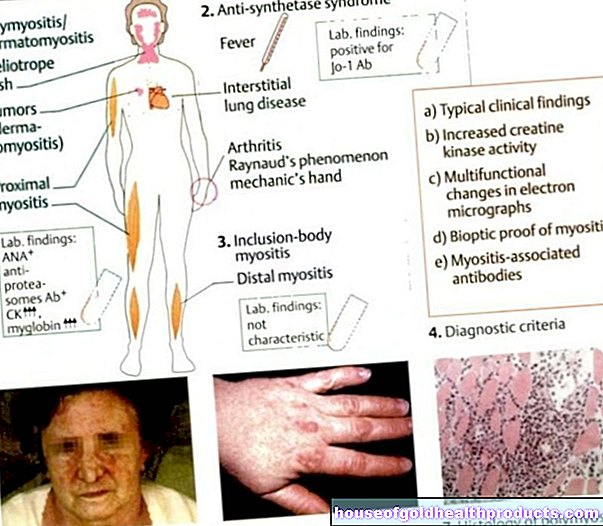










.jpg)