Pulmonal hypertensjon
Alt -innhold kontrolleres av medisinske journalister.Pulmonal hypertensjon (PH), også kjent som pulmonal arteriell hypertensjon (PAH) eller pulmonal hypertensjon, er en sykdom som får blodtrykket i lungesirkulasjonen til å stige kronisk (pulmonal hypertensjon). Blodårene i lungene er innsnevret, noe som fører til økt motstand i karene og dermed økt blodtrykk. Pulmonal hypertensjon er vanligvis forårsaket av en kronisk sykdom i hjertet eller lungene. Her finner du ut hvordan pulmonal hypertensjon kan utvikle seg og hvordan den behandles.
ICD -koder for denne sykdommen: ICD -koder er internasjonalt anerkjente koder for medisinske diagnoser. De finnes for eksempel i legebrev eller på attester om arbeidsuførhet. I27
Pulmonal hypertensjon: beskrivelse
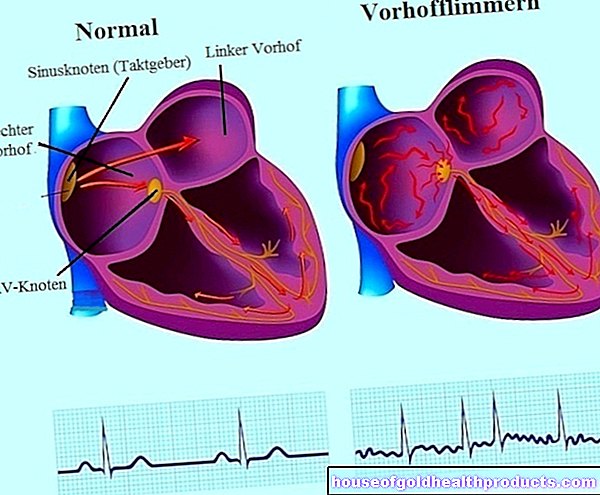
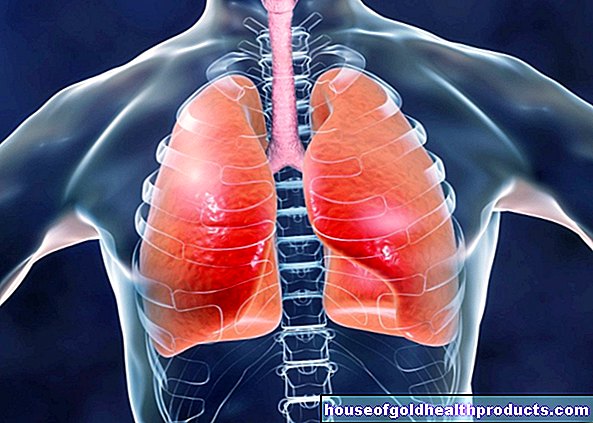
Pulmonal hypertensjon (forkortet PH) er en sykdom der lungekarene blir innsnevret på grunn av forskjellige årsaker. Dette øker blodtrykket i lungesirkulasjonen - dette er også kjent som pulmonal hypertensjon.
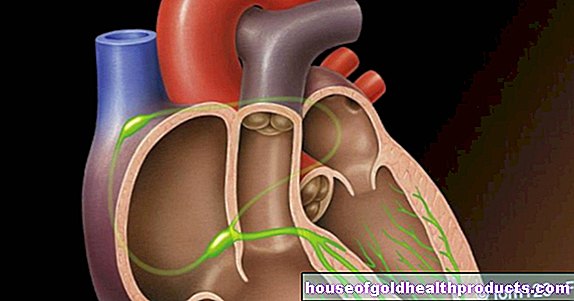
Følgende skjer i lungesirkulasjonen, også kjent som den lille sirkulasjonen: Blodet transporteres fra hjertet til lungene og tilbake igjen. Det oksygenfattige blodet når lungene fra høyre ventrikkel via lungestammen, som er delt inn i høyre og venstre lungearterie. Der er den igjen beriket med oksygen.
De innsnevrede karene ved pulmonal hypertensjon øker motstanden i lungene. Dette gjør det vanskelig for høyre ventrikkel å pumpe blod gjennom lungene. Resultatet: blodstrømmen gjennom lungearteriene blir forstyrret og blodtrykket økes, høyre ventrikel blir stadig mer overbelastet. Ytterligere konsekvenser kan være en sirkulasjonsforstyrrelse i lungene og dårlig oksygenopptak i lungene. I de mest alvorlige tilfellene fører pulmonal hypertensjon til hjertesvikt.
Pulmonal hypertensjon forårsaker ofte ikke symptomer på et tidlig stadium. Bare når sykdommen utvikler seg, vises symptomer. Siden kroppen ikke lenger er tilstrekkelig tilført oksygen ved pulmonal hypertensjon, er de som er rammet sterkt begrenset i fysisk ytelse, blir raskt utslitt og klager blant annet over symptomer som kortpustethet. Hvis den ikke behandles, kan pulmonal hypertensjon være dødelig.
I medisin skilles det mellom primær og sekundær pulmonal hypertensjon: I svært sjeldne tilfeller oppstår pulmonal hypertensjon som en uavhengig sykdom (f.eks. Gjennom arvelighet), så kalles det primær eller idiopatisk pulmonal hypertensjon (IPAH for kort). Som regel utløses imidlertid pulmonal hypertensjon av kroniske sykdommer, visse medisiner eller medisiner.Så er det snakk om sekundær pulmonal hypertensjon.
Tidlig påvisning av den underliggende sykdommen er spesielt viktig i terapien, slik at pulmonal hypertensjon ikke går videre. Pulmonal hypertensjon behandles vanligvis med medisiner for å redusere pulmonal hypertensjon.
Pulmonal hypertensjon: symptomer
Med pulmonal hypertensjon er symptomer ofte fraværende i de tidlige stadiene av sykdommen. Bare etter hvert som sykdommen utvikler seg, opplever de berørte symptomene. Den reduserte oksygentilførselen til lungene begrenser deres ytelse sterkt. Typiske symptomer på pulmonal hypertensjon er:
- Ineffektivitet
- Rask tretthet
- kortpustethet
- svimmelhet
- Mulig plutselig, kort bevissthetstap (synkope) under fysisk anstrengelse
- Blåaktig misfarging av hud og lepper (cyanose)
- Brystsmerter
- Hevelse på grunn av opphopning av væske i vevet (ødem), spesielt i beina
På den ene siden svekker de innsnevrede karene oksygentilførselen til lungene, på den annen side trenger hjertet mer og mer styrke for å pumpe blodet gjennom karene. Hjertet må slå raskere, og dette resulterer i en akselerert puls, noe som også kan merkes ved hjertearytmier. Hjertet blir stadig mer stresset, spesielt høyre atrium og høyre ventrikkel påvirkes. Høyre hjertesvikt (høyre hjertesvikt) kan utvikle seg.
Pulmonal hypertensjon: årsaker og risikofaktorer
Pulmonal hypertensjon er definert som en økning i gjennomsnittlig blodtrykk i lungearterien, det såkalte pulmonal arterielle trykket (PAPm). Hos en frisk person er gjennomsnittlig lungetrykk i hvile under 20 mmHg. Pulmonal hypertensjon er tilstede hvis verdien stiger over 25 mmHg i hvile og over 30 mmHg under trening.
Årsaken til pulmonal hypertensjon er en innsnevring av lungekarene, noe som reduserer karvolumet og reduserer blodstrømmen. Årsaken til vasokonstriksjonen er en ubalanse mellom visse messenger -stoffer som regulerer vasodilatasjon eller vasokonstriksjon. Årsaken til denne ubalansen er så langt ukjent. Celler i den indre vaskulære veggen (endotelceller), det indre laget og de glatte vaskulære musklene i lungearteriene blir i økende grad utsatt for vasokonstriktive messenger -stoffer (for eksempel endotelin, serotonin, tromboxan). Samtidig er vasodilaterende budbringersubstanser (som prostacyklin eller nitrogenoksid) tilstede i redusert antall. Som et resultat kontrakterer fartøyene. De vasodilaterende stoffene fører også til økt cellevekst, slik at karveggene tykner og karene smalner mer og mer. Disse prosessene reduserer karvolumet: mindre blod kan strømme gjennom dem; samtidig øker det lavere volumet også motstanden, noe som øker trykket som blodet strømmer gjennom karene. Dette øker også belastningen på hjertet. På grunn av den høyere motstanden i karene, må den bruke mer og mer kraft for å pumpe blod inn i lungesirkulasjonen. Over tid forstørres høyre hjertekammer og muskelen i hjerteveggen tykner til hjertet ikke lenger er i stand til å pumpe nok blod. Dette kan føre til såkalt høyre hjertesvikt.
Leger skiller mellom primær og sekundær pulmonal hypertensjon. Primær eller idiopatisk pulmonal hypertensjon (IPAH) forekommer som et uavhengig klinisk bilde uten en kjent årsak, så det er ingen underliggende sykdom som årsak. IPAH kan arves: Hvis to eller flere familiemedlemmer er påvirket i en familie, kalles det familiær pulmonal hypertensjon (FPAH). Imidlertid er begge former for pulmonal hypertensjon ekstremt sjeldne: Antallet nye tilfeller av begge former kombinert er bare ett til tre tilfeller per million innbyggere per år.
Vanligvis er en bestemt underliggende sykdom årsaken til pulmonal hypertensjon. Så er det snakk om sekundær pulmonal hypertensjon; pulmonal hypertensjon er derfor et resultat av den underliggende sykdommen. Spesielt pasienter med venstre hjertesvikt eller kroniske lungesykdommer lider ofte av pulmonal hypertensjon. Sykdommer som kan forårsake pulmonal hypertensjon inkluderer:
- KOLS (kronisk obstruktiv lungesykdom): Det er den vanligste årsaken til pulmonal hypertensjon.
- Lungefibrose: Ved denne sykdommen i lungevevet dannes mer bindevev, noe som blant annet fører til redusert oksygenopptak.
- Bindevevssykdommer som det såkalte CREST-syndromet eller sklerodermi
- Lungeemboli (blokkering av blodårer i lungene)
- HIV -infeksjon
- Venstre hjertesykdom
- Leversykdom
- Schistosomiasis (schistosomiasis): Denne orminduserte sykdommen er en vanlig årsak til pulmonal hypertensjon, spesielt i Sør-Amerika.
Enkelte medisiner som appetittundertrykkende midler (anorektika) og rusmisbruk anses også som risikofaktorer som kan forårsake pulmonal hypertensjon.
Avhengig av årsaken er pulmonal hypertensjon delt inn i fem kategorier:
- Pulmonal arteriell hypertensjon (PAH)
- Pulmonal hypertensjon ved venstre hjertesykdom
- Pulmonal hypertensjon ved lungesykdommer og / eller hypoksemi (lave nivåer av oksygen i arterielt blod)
- Pulmonal hypertensjon på grunn av kroniske trombotiske og / eller emboliske sykdommer
- Pulmonal hypertensjon på grunn av andre uklassifiserte sykdommer
Pulmonal hypertensjon: undersøkelser og diagnose
Pulmonal hypertensjon er ofte vanskelig å diagnostisere i begynnelsen fordi symptomene er ganske uspesifikke og også forekommer ved andre sykdommer. En detaljert diskusjon av sykehistorien (anamnese) og en fysisk undersøkelse gir viktig informasjon. Hvis det er mistanke om pulmonal hypertensjon, kan leger bruke forskjellige metoder for å undersøke hjerte og lunger:
- En ultralydundersøkelse (ekkokardiografi) av hjertet er den viktigste undersøkelsesmetoden for pulmonal hypertensjon. Dette gjør at legen kan undersøke størrelsen og funksjonen til hjertet og undersøke hjerteveggen og hjerteventilenes bevegelser. I tillegg er det mulig for ham å vise blodstrømmen inne i hjertet og registrere hastigheten på blodstrømmen. På denne måten kan han estimere et forhøyet blodtrykk i lungearteriene (pulmonal arterielt systolisk trykk).
- Med en røntgenundersøkelse av brystet kan leger identifisere forstørrede lungearterier (lungearterier). Disse er imidlertid bare gjenkjennelige på et avansert stadium av sykdommen, så en røntgenundersøkelse i begynnelsen av pulmonal hypertensjon er ikke veldig meningsfull.
- Et elektrokardiogram (EKG) er også viktig. Denne metoden måler hjertets elektriske aktivitet: legen mottar for eksempel informasjon om hjerterytmen og pulsen. Hvis pulmonal hypertensjon er tilstede, kan karakteristiske endringer sees i EKG.
- En lungefunksjonstest utføres også for å diagnostisere pulmonal hypertensjon. Den såkalte spirometrien brukes til å måle volumet i lungene eller pusten: I denne undersøkelsen puster pasienten inn i en spesiell enhet, spirometeret, som måler luftmengden som strømmer gjennom. Denne undersøkelsen brukes til å vurdere alvorlighetsgraden, prognosen og forløpet av pulmonal hypertensjon. Det kan også gi ledetråder om årsaken til pulmonal hypertensjon.
- For å bekrefte diagnosen pulmonal hypertensjon og for å bestemme alvorlighetsgraden av sykdommen, er en såkalt høyre hjertekateterundersøkelse egnet. Det kan brukes til å måle pulmonal arterielt blodtrykk direkte.
Den såkalte seks minutters gåtesten gir informasjon om graden av fysisk motstandskraft ved pulmonal hypertensjon. Den måler avstanden en pasient kan tilbakelegge på seks minutter i et behagelig tempo.
Når diagnosen pulmonal hypertensjon er gjort, er alvorlighetsgraden delt inn i ett av fire alvorlighetsgrader:
- Klasse 1: Ingen begrensninger på fysisk aktivitet. Normal belastning fører ikke til ubehag.
- Klasse 2: Liten begrensning av fysisk aktivitet, ingen klager i ro. Normal fysisk aktivitet fører til økt andpustenhet eller tretthet, besvimelse eller brystsmerter.
- Klasse 3: Betydelige begrensninger i fysisk aktivitet, ingen klager i ro. Lette aktiviteter forårsaker ubehag.
- Klasse 4: Pasienter med pulmonal hypertensjon av denne alvorlighetsgraden kan ikke utføre fysisk anstrengelse uten symptomer. I tillegg er det en høyre hjertesvakhet, andpustenhet og / eller tretthet oppstår selv i hvile. Den minste aktiviteten fører til en forverring av symptomene.
Pulmonal hypertensjon: behandling
Pulmonal hypertensjon behandler enten den underliggende tilstanden som forårsaker pulmonal hypertensjon eller behandler symptomene den forårsaker. Fordi en kur mot pulmonal hypertensjon ikke er mulig. Målet er å forlenge levealderen så vel som å forbedre fysisk motstandskraft og livskvalitet.
Pulmonal hypertensjon behandles vanligvis med medisiner. Avhengig av årsaken til sykdommen, brukes antihypertensive eller vasodilaterende midler. Disse inkluderer for eksempel:
- Kalsiumkanalblokkere med høy dose: Disse stoffene senker blodtrykket i lungene, men brukes bare hos pasienter med idiopatisk pulmonal hypertensjon (IPAH). I tillegg er deres effektivitet tidligere testet med et høyre hjertekateter.
- Prostacyklinderivater (prostanoider): De ligner kroppens eget messengerstoff prostacyklin og har en vasodilaterende effekt. Disse stoffene gis enten i en vene eller gjennom en inhalator.
- Fosfodiesterase (PDE) 5 -hemmere: Aktive ingredienser i denne gruppen senker blodtrykket i lungekarene.
- Såkalte endotelinreseptorantagonister motvirker kroppens eget messengerstoff endotelin, som virker vasokonstriktivt.
Hvis årsaken er kronisk obstruktiv lungesykdom (KOLS), kan pulmonal hypertensjon behandles med langvarig oksygenbehandling (med hjemmeventilasjon med maske) for å forbedre pustebesværet. Hvis medikamentell behandling for pulmonal hypertensjon ikke lykkes, er en hjerte-lungetransplantasjon ofte det siste alternativet.
Det finnes ingen standardbehandling for pulmonal hypertensjon - det avhenger av den underliggende sykdommen og alvorlighetsgraden av pulmonal hypertensjon. Behandlingen er derfor individuelt tilpasset pasienten.
Pulmonal hypertensjon: forebygging
Siden pulmonal hypertensjon bare forekommer i ekstremt sjeldne tilfeller som en uavhengig sykdom, men som en konsekvens av kroniske sykdommer, er det viktig å behandle det på et tidlig stadium. Dette er den eneste måten å forhindre pulmonal hypertensjon. Regelmessige kontroller med legen din er derfor avgjørende, spesielt hvis du allerede har en sykdom som anses som en risikofaktor for pulmonal hypertensjon.
Personer med pulmonal hypertensjon bør unngå tung fysisk anstrengelse. Dette gjelder spesielt for anstrengende sportsaktiviteter som ikke er under tilsyn av en lege, fordi de kan føre til ytterligere økning i pulmonal hypertensjon. Imidlertid kan fysisk opplæring fysisk trening forbedre tilstanden til mange pasienter og være et nyttig tillegg til terapi.
I tillegg anbefales pasienter med pulmonal hypertensjon ikke å reise til høyder over 2000 meter. fordi opphold i slike høyder kan forverre tilstanden. Flyreiser er derfor også en potensiell risiko på grunn av pulmonal hypertensjon.
Pulmonal hypertensjon: sykdomsforløp og prognose
Pulmonal hypertensjon skyldes hovedsakelig kroniske sykdommer i lungene eller hjertet. Det finnes ingen kur mot pulmonal hypertensjon - i verste fall fører det til riktig hjertesvikt. Behandling kan øke forventet levealder og generell livskvalitet.
Prognosen for pulmonal hypertensjon avhenger også av hvor høyt blodtrykket i lungene allerede er-jo høyere arterielt pulmonal trykk (PAPm), desto dårligere er den såkalte 5-års overlevelsesraten: Jo høyere PAPm, jo verre er det er -Verdier over 30 mmHg. Hvis det ikke er noen behandling i det hele tatt, er gjennomsnittlig levetid etter diagnose maksimalt tre år.
Tags.: Diagnose Sykdommer tenåring