Cystisk fibrose
og Christiane Fux, medisinsk redaktør og Martina Feichter, medisinsk redaktør og biologSophie Matzik er frilansskribent for det medisinske teamet til
Mer om -eksperteneChristiane Fux studerte journalistikk og psykologi i Hamburg. Den erfarne medisinske redaktøren har skrevet magasinartikler, nyheter og faktatekster om alle tenkelige helseemner siden 2001. I tillegg til arbeidet for, er Christiane Fux også aktiv i prosa. Hennes første kriminalroman ble utgitt i 2012, og hun skriver, designer og publiserer også sine egne krimspill.
Flere innlegg av Christiane FuxMartina Feichter studerte biologi med et valgfag apotek i Innsbruck og fordypet seg også i en verden av medisinske planter. Derfra var det ikke langt til andre medisinske emner som fortsatt fengsler henne den dag i dag. Hun utdannet seg til journalist ved Axel Springer Academy i Hamburg og har jobbet for siden 2007 - først som redaktør og siden 2012 som frilansskribent.
Mer om -ekspertene Alt -innhold kontrolleres av medisinske journalister.
Cystisk fibrose er en medfødt metabolsk sykdom. Hos de berørte er kroppsvæsker som spytt, bronkial slim eller bukspyttkjertelsekresjon mye tøffere enn vanlig. Dette forårsaker blant annet pusteproblemer og fordøyelsesforstyrrelser. Det er ingen kur mot cystisk fibrose. Med konsekvent terapi kan imidlertid sykdomsforløpet bremses. Les her om symptomene på cystisk fibrose og hvordan du behandler dem.
ICD -koder for denne sykdommen: ICD -koder er internasjonalt anerkjente koder for medisinske diagnoser. De finnes for eksempel i legebrev eller på attester om arbeidsuførhet. E84
Cystisk fibrose: rask referanse
- Beskrivelse: arvelig metabolsk sykdom, forårsaker at det dannes tykt slim i lungene og andre organer
- Symptomer: pusteproblemer, tørr hoste, følsomhet for infeksjoner i lungene, manglende evne til å trives, fordøyelsesbesvær, alvorlig diaré, fettlever, redusert fruktbarhet
- Årsaker: arv av defekte gener som påvirker konsistensen av kroppsvæsker. Sykdommen bryter imidlertid bare ut hvis begge foreldrene gir et sykt gen videre til barnet (autosomal recessiv arv)
- Diagnostikk: blodprøve for immunreaktivt trypsin (IRT), pankreatittassosiert protein (PAP), svette test, genetisk test
- Behandling: slimfortykningsmidler, bronkodilatatorer, innånding, antibiotika for bakterielle luftveisinfeksjoner, kortison, CFTR-modulatorer, lungetransplantasjon
- Prognose: uhelbredelig, kurset avhenger sterkt av alvorlighetsgraden og tidspunktet for diagnosen, forkortet levealder
Cystisk fibrose: beskrivelse
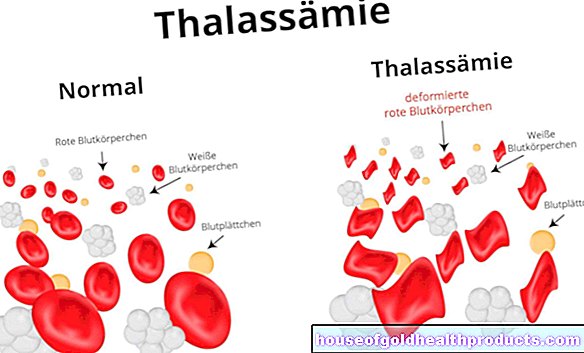
Cystisk fibrose (også kalt cystisk fibrose) er en arvelig metabolsk sykdom. Dannelsen av forskjellige kroppsvæsker forstyrres. Sekresjonen av lungene, bukspyttkjertelen og andre organer er mer tyktflytende enn hos friske mennesker.
Tykt slim
Det tøffe slimet tetter blant annet de små grenene av bronkiene og kanalene i de indre organene. Pust og fordøyelse er spesielt hardt påvirket. I løpet av sykdommen kan organene fungere mindre og mindre.
Dette er hva som skjer med cystisk fibrose
Feil i genomet
Årsaken til sykdommen er defekter i den genetiske sminken. Cystisk fibrose er derfor ikke herdbar. Tidspunkt for diagnose og alvorlighetsgraden av symptomer kan variere mye fra person til person. Hos mange barn er cystisk fibrose merkbar fra fødselen, i andre tilfeller gjenkjennes det først senere.
Cystisk fibrose: symptomer
Cystisk fibrose symptomer kan variere sterkt fra en pasient til en annen. Sykdommen påvirker funksjonen til forskjellige organer, men spesielt lungene og fordøyelsessystemet.
Kjenne igjen tidlige tegn
Når de første symptomene oppstår ved cystisk fibrose varierer fra person til person. I de fleste tilfeller oppstår symptomer på cystisk fibrose i løpet av det første leveåret. Dette betyr at sykdommen vanligvis kan diagnostiseres tidlig og behandling kan startes raskt. Noen pasienter har imidlertid bare klare symptomer i ungdomsårene. I tillegg viser ikke alle berørte personer hele spekteret av mulige symptomer. Alvorlighetsgraden av symptomene varierer også.
Endret kroppsvæske
Ved cystisk fibrose forstyrres dannelsen av de såkalte kloridionkanalene i cellemembranene. Dette endrer sammensetningen av kroppsvæsker. Den enkleste måten å bestemme denne endringen i svetten til de berørte. Svetten deres er mer salt enn hos friske mennesker. Saltene natrium og klorid, som er en del av de såkalte elektrolyttene, akkumuleres i svetten. Lider av cystisk fibrose mister flere kroppssalter gjennom svette.
Flere symptomer på cystisk fibrose
Sykdommen påvirker en rekke organsystemer. De første symptomene på cystisk fibrose vises ofte i lungene og fordøyelseskanalen. Ytterligere klager kan oppstå i løpet av livet. Symptomene kan behandles godt gjennom målrettet terapi. Imidlertid kan symptomene også være truende. Det er spesielt farlig når bronkialrørene er blokkert av det tykke slimet. Da kan pasientene i ekstreme tilfeller kveles.
-
"Sport kan lindre symptomer"
Tre spørsmål til
Suat Evcümen,
Spesialist i indremedisin og pulmonologi -
1
Hvorfor gjenkjennes cystisk fibrose så sent hos noen mennesker?
Suat EvcümenCystisk fibrose er en genetisk sykdom. Avhengig av hvor alvorlige mutasjonene er, er det også milde forløp som først blir merkbare sent. I utgangspunktet fungerer ikke visse kloridkanaler ordentlig, og derfor tykner pasientens kroppssekresjoner. Dette påvirker flere organsystemer, for eksempel luftveiene eller fordøyelsen. Det er derfor man snakker om en multisystem-sykdom.
-
2
Vil du anbefale nyfødt screening?
Suat EvcümenTidlig påvisning er ekstremt viktig og kan bidra til å påvirke sykdomsforløpet positivt gjennom tidlig behandling. Disse inkluderer for eksempel administrering av fordøyelsesenzymer, inhalasjonsterapi og tidlig fysioterapeutisk støtte. Av denne grunn har nyfødt screening blitt utført i Tyskland i en årrekke.
-
3
Hva betyr diagnosen?
Suat EvcümenForventet levetid for pasienter med cystisk fibrose har steget jevnt de siste to tiårene. Vi skylder dette til rettidig oppdagelse og tidlig oppstart av de individuelt tilpassede behandlingene. De fleste vokser opp med sykdommen og "lærer" å håndtere den. Vi fortsetter å observere at trening hjelper og lindrer symptomer. Søk også psykologisk hjelp.
-
Suat Evcümen,
Spesialist i indremedisin og pulmonologiSiden mars 2019 har Suat Evcümen vært overlege i pulmologi ved Paracelsus Clinic på Silbersee.

Cystisk fibrose: symptomer på lungene
Pustevansker og tørr hoste
I de fleste tilfeller av cystisk fibrose vises ikke lungesymptomer før litt eldre spedbarn. Nyfødte har vanligvis ingen pusteproblemer. Hos litt eldre barn har symptomene på cystisk fibrose ofte form av kikhostelignende, kronisk irritabel hoste. Slim i luftveiene er økt, tykt og tykt. Dette hindrer luftstrømmen i lungene. Over tid utvikler det seg pustevansker.
Hyppige infeksjoner
Den økte oppbyggingen av slim i lungene gjør det lettere for bakterier å bosette seg og forårsake en infeksjon. Tilbakevendende lungebetennelse eller bronkiale infeksjoner er hovedsakelig forårsaket av bakterier som stafylokokker og Pseudomonas -arter. Den forstyrrede saltbalansen i lungene hindrer også kroppens forsvar. Lungeblødning kan også forekomme. Et typisk tegn på dette er hoste opp slim blandet med blod.
Selv om lungene allerede er skadet fra tidlig barndom, vises de første symptomene på cystisk fibrose i luftveiene ofte bare i barneskolealderen eller enda senere. Symptomene er noen ganger bare merkbare når store deler av lungevevet allerede er ødelagt eller luftveiene er kraftig innsnevret.
Cystisk fibrose: symptomer på bukspyttkjertelen
Hos pasienter med cystisk fibrose blir bukspyttkjertelen ofte betent: Den skiller ut en sekresjon som blant annet inneholder enzymer for fordøyelse av fett og karbohydrater (eksokrin bukspyttkjertelfunksjon). Hos personer med cystisk fibrose bygger sekresjonen seg opp på grunn av viskositeten og forårsaker betennelse i bukspyttkjertelen (pankreatitt).
Etter hvert som prosessen skrider frem, herdes bukspyttkjertelen og blir arrdannet. Leger snakker om fibrose. Fibrose ødelegger bukspyttkjertelen gradvis.
I tillegg til fordøyelsessekresjonen, produserer bukspyttkjertelen også hormonet insulin (endokrin bukspyttkjertelfunksjon) i spesielle celler - betacellene. Blant annet er det nødvendig for kroppen å bruke sukker. Hos pasienter med cystisk fibrose ødelegger den viskøse fordøyelsessekresjonen i bukspyttkjertelen kanalene betacellene gradvis - insulinproduksjonen svekkes og insulinmangel utvikles (spesielt hos pasienter over 10 år). Som et resultat kan en form for diabetes (diabetes mellitus) utvikle seg: diabetes type 3.
Cystisk fibrose: symptomer på galle
Bukspyttkjertelen og galleblæren deler en felles kanal i tarmen. Derfor kan etterslepet av bukspyttkjertelsekresjoner også forårsake betennelse i galleblæren. Gallestein dannes ofte også, noe som helt kan blokkere drenering av galle fra galleblæren.
Cystisk fibrose: symptomer på fordøyelseskanalen
I tillegg til klager fra lungene forårsaker cystisk fibrose ofte symptomer i fordøyelseskanalen. For eksempel påvirker mangelen på galle fettfordøyelsen. Som et resultat finner pasienter det ofte vanskelig å tåle fet mat. Mesteparten av maten som blir inntatt, skilles ut ufordøyd. Veldig voluminøs og myk avføring er typisk.
Diaré og hemmet vekst
Berørte barn, inkludert spedbarn, lider ofte av alvorlig diaré. Selv om de drikker og spiser nok, går de neppe opp i vekt. Stunted vekst og underernæring er derfor ytterligere klassiske konsekvenser av sykdommen.
Slik fordøyelsesbesvær kan også forekomme med mange andre sykdommer. Bare i kombinasjon med pusteproblemer er de en indikasjon på cystisk fibrose, som du definitivt bør undersøke.
Anal prolaps
Etter hvert som det utvikler seg, kan cystisk fibrose føre til forskjellige komplikasjoner i fordøyelseskanalen. Det vanligste er det som kalles anal prolaps. En del av slimhinnen i anus buler ut av anus. En slik hendelse må opereres så snart som mulig.
Tarmobstruksjon
En invagasjon av tarmen (intussusception) eller en intestinal obstruksjon (ileus) forekommer også ofte. Begge komplikasjonene er ledsaget av alvorlige magesmerter og betydelige fordøyelsesproblemer. Smerten oppstår vanligvis i anfall, spesielt etter å ha spist. En tarmobstruksjon er dødelig hvis den ikke behandles. Krampaktig, akutt magesmerter bør derfor alltid avklares av lege.
Cystisk fibrose: symptomer på leveren
Fettlever
Leveren lider også av etterslepet av galle. Fet lever utvikler seg hos mange pasienter etter hvert som sykdommen utvikler seg. Dette kan ledsages av tretthet, tap av matlyst, metthet og flatulens, og i sjeldne tilfeller trykkfølelse eller lette smerter i øvre del av magen.
Krympe leveren
I sjeldne tilfeller utvikler pasienter med cystisk fibrose krympet lever (skrumplever). Denne alvorlige leverdysfunksjonen blir først merkbar i form av gulsott (gulsott). En gulaktig misfarging av det hvite i øynene er et tegn på gulsott. Hjerteproblemer oppstår også etter en lang periode, og ytelsen til de berørte fortsetter å synke.
Cystisk fibrose: Redusert fruktbarhet
Mer enn halvparten av alle mannlige cystisk fibrose -pasienter er infertile. I de fleste tilfeller produserer de berørte befruktbare sædcellene i testiklene. Disse kan imidlertid ikke komme gjennom vas deferens fordi de er blokkert av tyktflytende slim.
Kvinner med cystisk fibrose er vanligvis mindre fruktbare. I prinsippet kan de bli gravide og bære et barn. Imidlertid akkumuleres tøft slim i egglederne, som sædcellene neppe kan trenge gjennom. Sannsynligheten for graviditet reduseres raskt, spesielt når folk blir eldre.
Cystisk fibrose: symptomer hos barn
Cystisk fibrose er en genetisk sykdom. Den har derfor alltid vært tilstede siden fødselen. Men de klassiske symptomene på cystisk fibrose vises ikke alltid i barndommen. Imidlertid er det ofte uspesifikke tips som bør følges opp. Dette gjelder spesielt hvis familien allerede har hatt tilfeller av cystisk fibrose.
Symptomer i fordøyelseskanalen
En indikasjon på en metabolsk lidelse er for eksempel en oppblåst mage i lang tid. Barna lider ofte av diaré. Hos nyfødte kan en veldig forsinket første tarmbevegelse (Kindspech) være en indikasjon på cystisk fibrose. I mange tilfeller oppstår det manglende evne til å trives og stunt, selv om barna spiser med voldsom appetitt. Forstoppelse som følge av sykdommen forekommer bare i sjeldne tilfeller.
Symptomer i luftveiene
Andre symptomer som kan indikere cystisk fibrose er raslende pust og alvorlig rastløshet. Mange barn lider av kronisk sinusbetennelse. Dette er spesielt merkbart gjennom smerter i ansiktet som ikke kan lokaliseres nøyaktig. Nesepolypper er også mer vanlig hos barn med cystisk fibrose enn hos friske barn.
Hvis barn lider av pusteproblemer eller fordøyelsesbesvær i lang tid, er det alltid lurt å oppsøke lege som et forebyggende tiltak. Livstruende situasjoner kan raskt oppstå hos barn fordi de ikke kan formulere klagene sine selv eller ikke kan vurdere alvorlighetsgraden.
Cystisk fibrose: årsaker og risikofaktorer
Cystisk fibrose er forårsaket av en genetisk defekt. Den patologiske endringen er på det syvende kromosomet i det såkalte CFTR-genet.
CFTR -genet (cystisk fibrose transmembranregulatorgen) inneholder byggeinstruksjonene for en kanal i cellemembranen som kloridioner kommer inn i cellen. De defekte kloridionkanalene blokkerer transport av salt til visse kroppsceller hos pasienter med cystisk fibrose.
De berørte kjertelcellene skiller deretter ut hardt slim i stedet for ellers flytende sekresjon. I lungene, paranasale bihuler, bukspyttkjertelen, i tarmen, i galleveiene og i gonadene dannes tøffe slimhinner med høyt saltinnhold.
Cystisk fibrose: hvor utsatt er barnet mitt?
Cystisk fibrose bryter først ut når begge foreldrene gir et patologisk endret CFTR -gen til barnet sitt. Disse foreldrene er vanligvis begge friske selv - de har et sunt og defekt CFTR -gen.
Personer med cystisk fibrose er bare delvis fruktbare. Noen pasienter blir fortsatt foreldre. Syke fedre eller mødre gir alltid et sykt gen videre, siden begge CFTR -genene bærer informasjon om cystisk fibrose. Barna dine blir imidlertid bare syke hvis de også har mottatt et sykt gen fra den andre forelderen.
Par som har hatt tilfeller av cystisk fibrose i familien, bør søke genetisk rådgivning før de planlegger en graviditet.
Slik arves cystisk fibrose
Preimplantasjonsdiagnostikk
Foreldre som kan gi cystisk fibrose videre til barnet sitt, kan dra fordel av diagnostikk før implantasjon. Eggcellene blir først kunstig befruktet. De første celledelingene finner sted i reagensrøret (in vitro).
Før et embryo opprettet på denne måten settes inn i kvinnens livmor, kontrolleres det for endrede genetiske egenskaper.Da blir det bare implantert embryoer som ikke inneholder det syke genet.
Uavhengig av dette kan det også undersøkes under graviditeten om barnet senere vil utvikle cystisk fibrose.
Cystisk fibrose: undersøkelser og diagnose
I motsetning til for noen få år siden, sjekker de fleste sykehus nå rutinemessig for nyfødte. Det inkluderer blant annet en undersøkelse for cystisk fibrose. Deltakelse i denne screeningsprosessen er frivillig og krever samtykke fra foreldrene til den nyfødte.
Screening i blod og svette
Blod hentes fra den nyfødte for screening. Testen for cystisk fibrose har flere stadier:
- Blodprøve: test for økte nivåer av immunreaktivt trypsin (IRT) og pankreatittassosiert protein (PAP). Hvis det er abnormiteter, utføres svette -testen.
- Svette test: Personer med cystisk fibrose har et betydelig høyere saltinnhold i svetten enn friske mennesker. For svette -testen for cystisk fibrose måles innholdet av saltene natrium og klorid i kroppssvette. Hos barn samles svette på underarmen og analyseres deretter. Hvis det oppstår mistanke her, utføres en genetisk test.
Genetisk test
Hos pasienter med cystisk fibrose endres det såkalte CFTR-genet, som gir bygningsinstruksjoner for visse ionekanaler. Disse instruksjonene er lange, de består av rundt 6500 basepar. En feil kan krype inn i koden hvor som helst - cystisk fibrose kan derfor være basert på forskjellige genetiske endringer (mutasjoner). Disse har forskjellige effekter. Derfor testes bare de viktigste avvikene i CFTR -genet.
Hvis begge CFTR -genene, det til faren og moren, viser en mutasjon, bekrefter dette - sammen med den positive svette -testen - diagnosen cystisk fibrose.
Familiehistorie gir ledetråder
Hvis det ikke er utført passende nyfødt screening og mistanken om cystisk fibrose oppstår senere, er familielegen eller internisten den rette kontakten. I en første samtale registrerer han sykehistorien (anamnese). Hvis det er mistanke om cystisk fibrose, er hovedfokuset på familiehistorie (er det noen kjente cystiske fibrose -sykdommer i familien? Er en av foreldrene kjent for å være bærer av et defekt CFTR -gen?).
Undersøkelser
En fysisk undersøkelse vil da finne sted. Legen lytter til pasientens lunger og skanner de indre organene. På denne måten kan han allerede utelukke noen andre sykdommer som er forbundet med symptomer som ligner på cystisk fibrose.
Svette -testen og gentesten, som beskrevet ovenfor, hjelper også hvis det er mistanke om cystisk fibrose hos eldre barn, ungdom eller voksne.
Når diagnosen er stilt, kan legen bruke forskjellige undersøkelser for å kontrollere i hvilken grad indre organer er nedsatt i funksjonen, og om komplikasjoner eller sekundære sykdommer allerede dukker opp. Slike undersøkelser utføres noen ganger rutinemessig for å overvåke utviklingen av cystisk fibrose.
Blodprøver kan for eksempel brukes til å avgjøre om insulinsekresjonen fra bukspyttkjertelen reduseres og blodsukkernivået økes - symptomer på diabetes. Leverfunksjonsverdier og konsentrasjonen av fettløselige vitaminer kan også måles i blodet.
Konsentrasjonen av fordøyelsesenzymet elastase (kommer fra bukspyttkjertelen) og fettkonsentrasjonen bestemmes i avføringsprøver.
Vevsprøver fra pasientens hals eller sputum undersøkes for bakterielle infeksjoner i laboratoriet.
Imaging prosedyrer (for eksempel røntgenstråler, computertomografi = CT) er også ofte avslørende. De kan for eksempel brukes til å oppdage lungeinfeksjoner. Nesepolypper kan også påvises ved bruk av CT.
Lungefunksjonsmålinger (lungefunksjonstester) viser hvor godt lungene fungerer.
Testing av familiemedlemmer
Hvis cystisk fibrose blir funnet i en familie, er det fornuftig at alle andre familiemedlemmer også gjennomgår en undersøkelse. Cystisk fibrose kan også forekomme i milde former. Da tar det ofte mange år før cystisk fibrose blir merkbar med tydelige symptomer. Ikke desto mindre er tidlig diagnose og cystisk fibrose -behandling også viktig for disse pasientene for å øke levealderen.
Cystisk fibrose: behandling
Det er ingen kur mot cystisk fibrose. Barn født med cystisk fibrose vil lide av sykdommens effekter resten av livet. Med en kombinasjon av fysioterapi, medisiner og innånding kan sykdomsforløpet bremses betydelig. Hovedmålet med cystisk fibrose -terapi er å sette de berørte i stand til å leve et så normalt liv som mulig.
Lære å leve med sykdommen
Det er spesielt viktig for barn å lære å håndtere sykdommen på lang sikt. Barn med cystisk fibrose bør lære hva sykdommen betyr og hvordan den påvirker kroppen så tidlig som mulig. Et sykehusopphold med spesielle opplæringsenheter kan være nyttig for dette. Barn og foreldre lærer hvordan de skal spise, hvordan sport er og hvordan man best oppfører seg i kritiske situasjoner. Foreldre og barn er også informert om hygienetiltak (i henhold til anbefalingene fra Robert Koch Institute): vanlig og omfattende håndhygiene (håndvask med såpe og vann), ingen polstrede møbler og potteplanter.
Hjelp til lungene
Det er forskjellige alternativer for behandling av symptomene på cystisk fibrose. Hvilke av disse som er nyttige i enkeltsaker avhenger av pasientens alder og alvorlighetsgraden av symptomene.
Ekspektoranter
Cystisk fibrose lider mest av lungeproblemer. Regelmessig innånding med spesielle tilsetningsstoffer (mukolytiske midler) løsner det tøffe slimet og gjør det lettere å hoste opp.
Bronkodilatatorer
Såkalte beta-2 sympatomimetika utvider bronkiene, noe som gjør pusten enda enklere.
Medisiner mot luftveisinfeksjoner
Dårlig ventilasjon av lungene gjør mennesker med cystisk fibrose mer utsatt for bakterielle luftveisinfeksjoner. De berørte bør behandles med antibiotika på et tidlig stadium. Den behandlende legen foreskriver ofte antibiotika som skal inhaleres, fordi de aktive ingrediensene når målstedet (lungene) direkte. Alternativt eller i tillegg kan inntak av antibiotika (f.eks. I form av tabletter) være indikert.
Ved alvorlige infeksjoner må antibiotika ofte gis i venen (intravenøst). Dette kan være nødvendig ved for eksempel kroniske infeksjoner med Pseudomonas aeruginosa.
Sopp luftveisinfeksjoner kan behandles med soppdrepende midler (antimykotika). Virushemmende legemidler (antivirale midler) kan ofte hjelpe mot virale luftveisinfeksjoner.
Antiinflammatoriske legemidler
Hos mange pasienter er luftveiene ofte eller kronisk betent. Deretter brukes antiinflammatoriske legemidler, spesielt glukokortikoider (kortison). De kan inhaleres, tas gjennom munnen eller gis i en vene.
CFTR -modulatorer
I noen år har såkalte CFTR-modulatorer vært tilgjengelige for behandling av cystisk fibrose. Disse stoffene, tatt på lang sikt, kan forbedre funksjonen til ionekanalene som er defekte av CFTR-mutasjonen. Dette kan forbedre pasientens lungefunksjon, øke vekten, senke saltinnholdet i svette og redusere hyppigheten av lungeinfeksjoner.
Dette er imidlertid mutasjonsspesifikke terapier, noe som betyr: CFTR-modulatorer fungerer bare med visse mutasjoner og derfor bare noen gang for visse pasienter. Noen eksempler:
Ivacaftor ble godkjent som den første CFTR -modulatoren i 2012. Det kan forskrives til babyer fra 12 måneder og som veier minst syv kilo hvis de har minst en såkalt gating-mutasjon (for eksempel G551D).
Cystisk fibrose -pasienter 12 år og eldre som har to kopier av F508del -mutasjonen kan behandles med en kombinasjon av de to CFTR -modulatorene ivacaftor og lumacaftor.
Forskere jobber intensivt med andre legemidler som vil hjelpe mot andre mutasjoner som ligger til grunn for cystisk fibrose.
Fysioterapier
Ulike fysioterapiteknikker hjelper til med å frigjøre luftveiene fra tøffe bronkiale sekresjoner. Disse inkluderer dreneringslagring, vibrasjonsdrenering, tappemassasje og hostetrening. Foreldre til små barn med cystisk fibrose kan lære disse teknikkene for pusteterapi og bruke dem daglig hjemme. Eldre barn og voksne med cystisk fibrose kan utføre åndedrettsterapi uavhengig (etter instruksjon fra en terapeut).
Lungetransplantasjon - det siste håpet
Hvis cystisk fibrose er alvorlig, er en lungetransplantasjon et alternativ. Så drastisk som dette trinnet kan virke i begynnelsen, kan mange pasienter da leve et liv med betydelig færre symptomer.
Spiser riktig for cystisk fibrose
Siden fordøyelsen forstyrres ved cystisk fibrose, må pasientene følge nøye med på kostholdet. Måltider bør gi nok kalorier og protein til å støtte normal vekst. Et normalt til høyt fettinntak anbefales også. Pasienter får fordøyelsesenzymer for å støtte matutnyttelse. Det finnes også preparater med fettløselige vitaminer (A, D, E, K) og mineraler. Sistnevnte erstatter saltene som pasientene svetter ut i store mengder.
En viss mengde bordsalt bør inntas sammen med mat hele dagen. Tilsvarende mer kreves ved høye temperaturer, feber, diaré, oppkast. En eventuell jernmangel bør testes en gang i året og erstattes ved mangel.
Spedbarn med cystisk fibrose
Babyer med cystisk fibrose bør bare mates med morsmelk de første fire månedene, så lenge de trives. Det er til og med bevis på at amming i mer enn seks måneder er forbundet med mindre alvorlighetsgrad. Forskning har vist at ammende spedbarn har bedre lungefunksjon og færre infeksjoner i løpet av de tre første leveårene enn spedbarn med cystisk fibrose som får morsmelkerstatning.
Hvis morsmelk ikke er mulig, bør spedbarn gis kommersiell formel når de trives.
Hvis babyer med cystisk fibrose ikke trives, anbefales morsmelkerstatning med høyt kaloriinnhold (i tillegg til morsmelk for ammende spedbarn).
Når det gjelder introduksjon av komplementær mat, gjelder de samme anbefalingene som for friske barn: foreldre bør begynne med fôring tidligst i begynnelsen av 5. måned og senest i begynnelsen av 7. måned.
Kostholdet til barn med cystisk fibrose bør tilpasses deres behov, slik at de minste kan trives i henhold til alderen. Både overvekt og undervekt bør unngås. Et kosthold av god kvalitet er også tilrådelig. Du bør være oppmerksom på gode diettfett (med enumettede og flerumettede fettsyrer, omega-3 fettsyrer).
Lungefunksjonen eller veksten av alveolene påvirkes av fysisk utvikling de første leveårene. Derfor bør barn med cystisk fibrose i denne aldersgruppen regelmessig undersøkes for ernæringsstatus (basert på målinger av vekt, kroppslengde og hodeomkrets).
Ta vaksinasjoner
Vaksinasjoner er spesielt viktige for personer med cystisk fibrose. Bakterier har det lettere med dem, og de er ofte mer syke enn pasienter som ikke er stresset på forhånd.
I utgangspunktet bør barn med cystisk fibrose motta alle vaksinasjoner som den stående vaksinasjonskommisjonen (STIKO) anbefaler for friske avkom. I tillegg anbefales en hepatitt A -vaksinasjon for barn i alderen 12 måneder og over, og en årlig influensavaksinasjon med tetravalent inaktivert influensavaksine for barn i alderen seks måneder og oppover. For babyer under seks måneder bør alle husstandsmedlemmer og omsorgspersoner vaksineres mot influensa for å beskytte barnet.
Barn med cystisk fibrose kan også gis et forebyggende legemiddel (palivizumab) mot RSV -patogenet (respiratorisk syncytialvirus). Disse virusene, som er spredt over hele verden, kan forårsake luftveissykdommer, spesielt hos babyer og småbarn.
Cystisk fibrose: sykdomsforløp og prognose
Cystisk fibrose er forårsaket av en endring i den genetiske sammensetningen og er derfor uhelbredelig. Generelt er forventet levetid og livskvalitet vanligvis betydelig redusert ved cystisk fibrose. Uten terapi forverres helsetilstanden raskt, og de som rammes lever vanligvis ikke lenge.
Med rettidig og konsistent terapi kan sykdomsforløpet bremses betraktelig. Pasientene lever nå betydelig lengre enn de gjorde for noen år siden. Gjennomsnittlig levealder med cystisk fibrose er for tiden rundt 50 år. Men mange pasienter blir også eldre.
Komplikasjoner og sekundære sykdommer
Selv med intensiv terapi kan komplikasjoner oppstå igjen og igjen med cystisk fibrose. Oftest oppstår akutt kortpustethet på grunn av dårlig lungeventilasjon. Individuelle områder av lungene kan til og med kollapse (atelektase).
Kronisk bronkitt eller lungebetennelse utvikler seg ofte. Sopp kan også påvirke lungene.
I tillegg kan endringer i væske- og elektrolyttbalansen utløse sjokk og sirkulasjonssvikt.
Andre mulige komplikasjoner og sekundære sykdommer ved cystisk fibrose er:
- kronisk leversykdom, spesielt skrumplever
- Betennelse i galleblæren og gallestein
- kronisk pankreatitt
- forstyrret hjertefunksjon
- akutt tarmobstruksjon (ileus)
- Tarminvagasjon (intussusception)
- Underernæring
- Sukkersyke
- redusert fruktbarhet hos kvinner og infertilitet hos menn
Cystisk fibrose: forebygging?
Cystisk fibrose er en arvelig sykdom, så forebygging er ikke mulig. Personer med økt familiær risiko bør søke genetisk rådgivning hvis de ønsker å få barn. En genetisk test utføres for å avgjøre om CFTR -genet er endret. Avhengig av om en eller begge partnerne bærer det defekte genet, kan risikoen for avkommet beregnes.
I mellomtiden er pre-implantasjonsdiagnose (PGD) mulig for cystisk fibrose i de fleste europeiske land i henhold til lovbestemmelsene i det respektive landet. Eggcellene blir befruktet utenfor livmoren og det brukes bare embryoer uten de problematiske genene for cystisk fibrose. Forutsetningen for dette er alltid godkjenning av en etisk komité.
Tilleggsinformasjon
Retningslinjer:
- S2 konsensusretningslinje "Diagnose av cystisk fibrose" fra Society for Pediatric Pneumology
- S3 retningslinje "Lungesykdom ved cystisk fibrose" fra Society for Pediatric Pneumology
- S3 retningslinje: "Cystisk fibrose hos barn i de to første leveårene, diagnostikk og terapi" fra Society for Pediatric Pneumology (GPP), German Society for Child and Adolescent Medicine (DGKJ) og andre
Selvhjelpsgrupper:
- Cystisk fibrose e.V.: Https://www.muko.info/
- CF Austria: Cystic Fibrosis Help Austria: https://www.cf-austria.at/
- Cystisk fibrose Sveits: https://cystischefibroseschweiz.ch/







.jpg)
-der-giraffentrick.jpg)