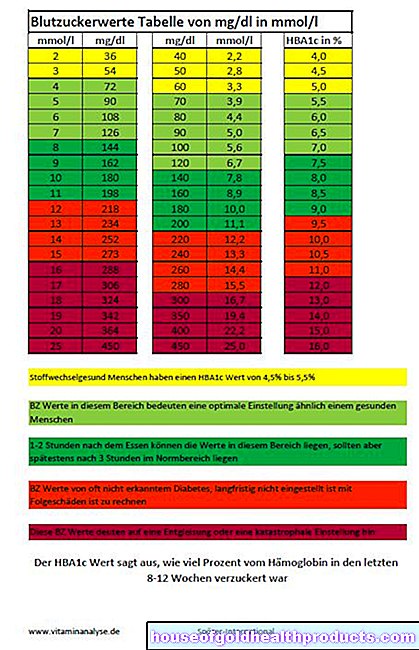
Cystisk fibrose: snart behandlingsbar i stedet for dødelig?
Christiane Fux studerte journalistikk og psykologi i Hamburg. Den erfarne medisinske redaktøren har skrevet magasinartikler, nyheter og faktatekster om alle tenkelige helseemner siden 2001. I tillegg til arbeidet for, er Christiane Fux også aktiv i prosa. Hennes første kriminalroman ble utgitt i 2012, og hun skriver, designer og publiserer også sine egne krimspill.
Flere innlegg av Christiane Fux Alt -innhold kontrolleres av medisinske journalister.Takket være nye kombinasjoner av aktive ingredienser, kan den alvorlige arvelige sykdommen cystisk fibrose snart lett behandles for 90 prosent av de berørte.
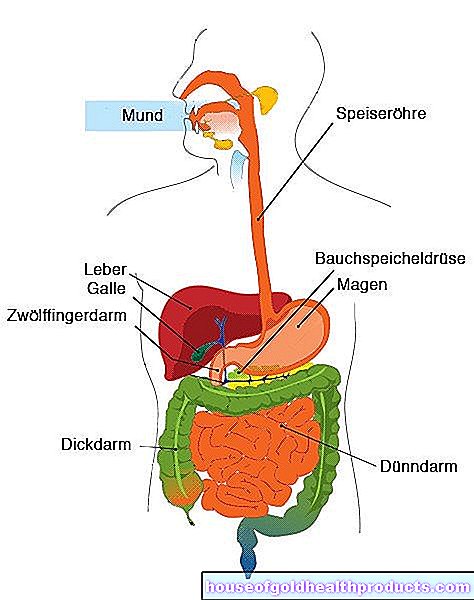
Cystisk fibrose er en (så langt) livsforkortende arvelig sykdom. Tøft slim dannes i forskjellige organer, inkludert lungene, og skader dem. I lang tid var det bare mulig å lindre symptomene og bremse sykdomsforløpet, men ikke å bekjempe årsaken - pasientene døde unge.
Gjennombrudd - men ikke for alle
Gjennombruddet kom med en kombinasjon av aktive ingredienser som kom på markedet for noen år siden. Det har fundamentalt endret cystisk fibrose -behandling og forbedret livskvaliteten og helsen til de som er rammet radikalt. Men det hjelper ikke alle pasienter: Ulike mutasjoner avbryter virkningsmekanismen. Nå er det håp for mange av dem også.
Kombinasjonen av aktive ingredienser tar sikte på å aktivere visse ionekanaler i pasientens cellemembraner, nærmere bestemt kloridkanalene. De regulerer transporten av salt og vann i slimhinner og hud. CFTR -genet er ansvarlig for dannelsen av kanalene.
Rettelser til kloridkanalen
Hos 90 prosent av pasientene i cystisk fibrose, på grunn av en spesiell mutasjon i CFTR -genet (F508del), brettes kloridkanalene feil under dannelsen, og derfor er det ikke engang cellen som inneholder dem i membranen. CFTR -korrektorer som elexacaftor og tezacaftor korrigerer feilfoldingen, som til slutt stabiliserer salt- og vannbalansen. De forskjellige sekresjonene i kroppen blir tynnere, oppløses lettere og hindrer ikke lenger lungene og andre organers arbeid.
Imidlertid blir rundt 15 prosent av pasientene også rammet av forskjellige andre mutasjoner. Disse har den effekten på forskjellige måter at kloridkanalene, selv om de er innebygd, ikke virker i det hele tatt eller fungerer i begrenset grad.
Dette er akkurat det som nå kan rettes opp eller lindres med en ny trippelkombinasjon av aktive ingredienser. Den består av elexacaftor, tezacaftor og også en såkalt CFTR-potensiator som ivacaftor, som effektivt aktiverer de innebygde kanalene.
Ni av ti pasienter kan få hjelp
"Dette betyr at vi i fremtiden effektivt vil kunne behandle rundt ni av ti mennesker med cystisk fibrose i Tyskland for den underliggende molekylære defekten," forklarer medlederforfatter prof. Marcus Mall, som blant annet er direktør for Christiane Herzog Cystic Fibrosis Center på Charité.
At trippelkombinasjonen er svært effektiv, er nå bekreftet i en klinisk fase 3 -studie med 258 pasienter med cystisk fibrose.
“Trippelterapien førte til en betydelig forbedring i CFTR -funksjon, lungefunksjon og livskvalitet sammenlignet med standarden på behandling. Også i disse pasientgruppene var trippelterapien - som i tidligere studier - også trygg og godt tolerert, sier Mall.
Hvis kombinasjonen av aktive ingredienser er godkjent, kan 90 prosent av pasientene med cystisk fibrose bli behandlet effektivt i fremtiden.
Tidlig behandling forhindrer langsiktig skade
Mall håper at selv spedbarn kan behandles: “Spesielt tidlig behandling av molekylære defekter som forårsaker sykdommen gjennom trippel terapi har et enormt potensial for å forhindre utvikling av irreversibel lungeskade som tidligere har begrenset pasientens levetid. Dette gjør cystisk fibrose fra en dødelig til en arvelig sykdom som kan behandles, forklarer forskeren. Minst for 90 prosent av pasientene.
Cystisk fibrose rammer én av 2500 til 3500 mennesker - vanligvis i tidlig barndom. Det er for tiden fortsatt den vanligste dødelige arvelige sykdommen.
Tags.: Sykdommer gpp kvinners helse