hemofili
og Martina Feichter, medisinsk redaktør og biologDr. med. Fabian Sinowatz er frilanser i det medisinske redaksjonen i
Mer om -eksperteneMartina Feichter studerte biologi med et valgfag apotek i Innsbruck og fordypet seg også i en verden av medisinske planter. Derfra var det ikke langt til andre medisinske emner som fortsatt fengsler henne den dag i dag. Hun utdannet seg til journalist ved Axel Springer Academy i Hamburg og har jobbet for siden 2007 - først som redaktør og siden 2012 som frilansskribent.
Mer om -ekspertene Alt -innhold kontrolleres av medisinske journalister.
Hemofili (blodsykdom) er en blodproppsforstyrrelse som vanligvis går i arv. De som lider mangler viktige blodkoagulasjonsfaktorer, eller de er defekte. Som et resultat er hemofile lett for blødninger og blåmerker. Hemofili er ennå ikke kurert. Takket være moderne terapier kan imidlertid hemofili føre et stort sett normalt liv. Les alt du trenger å vite om hemofili her.
ICD -koder for denne sykdommen: ICD -koder er internasjonalt anerkjente koder for medisinske diagnoser. De finnes for eksempel i legebrev eller på attester om arbeidsuførhet. D66D67D68
Kort overblikk
- Hva er hemofili? En genetisk blodproppsforstyrrelse (koagulopati). Det kalles også hemofili.
- Hemofili: Den vanligste er hemofili A, etterfulgt av hemofili B. Andre former som hemofili C, von Willebrand-Jürgens syndrom og parahemofili er mindre vanlige.
- Årsak: Mangel eller defekt i koagulasjonsfaktorer (blodproteiner som er nødvendige for at blodet skal størkne). Det er for det meste arvet og sjelden ervervet (forårsaket av en spontan genmutasjon).
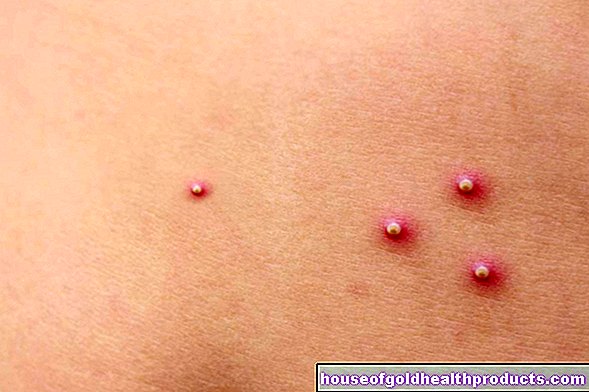
- Symptomer: økt tendens til blødning, som lett kan føre til blødninger og blåmerker (hematomer). Blødningen tar også lengre tid enn normalt. Hvor alvorlige symptomene er, avhenger av alvorlighetsgraden av hemofili.
- Diagnose: Måling av forskjellige blodparametere (aPTT, Hurtigverdi, plasmatrombintid, blødningstid, antall blodplater), bestemmelse av koagulasjonsfaktorers aktivitet
- Behandling: erstatning av den manglende koagulasjonsfaktoren (i form av faktorkonsentrater); i visse tilfeller andre legemidler (for eksempel desmopressin for mild hemofili A)
Hemofili: beskrivelse
Hemofili er en medfødt lidelse av blodpropp. De som rammes (hemofili, "hemofili") kan ikke danne tilstrekkelig funksjonelle koagulasjonsfaktorer. Dette er proteiner i blodet som er nødvendige for at blodet skal størkne. På grunn av mangel på koagulasjonsfaktorer kan det ikke lett dannes blodpropper og sår. Derfor er hemofili -pasienter utsatt for blødning. De har også sår som blør lenger enn normalt. Dette kan være farlig under visse omstendigheter.
Det medisinske uttrykket for en blodproppsforstyrrelse med økt tendens til blødning er "hemorragisk diatese". Det generelle navnet på en koagulasjonsforstyrrelse er "koagulopati".
Hemofili: frekvens
Hemofili forekommer nesten utelukkende hos gutter og menn. Det er relativt sjeldent: bare omtrent to av 10.000 menn har hemofili. Det er rundt 10.000 mennesker berørt i Tyskland. Rundt 3000 til 5000 av dem har en alvorlig form for sykdommen.
Hemofili: former
Det er forskjellige koagulasjonsfaktorer. Avhengig av hvilken koagulasjonsfaktor som påvirkes ved hemofili, skiller medisinske fagfolk mellom ulike former for sykdommen.
Hemofili A.
Ved hemofili A er det problemer med koagulasjonsfaktor VIII (antihemofilt globulin A): Enten kan kroppen ikke produsere den i tilstrekkelige mengder eller den er defekt. Omtrent 85 prosent av alle hemofili lider av hemofili A. Nesten alle er menn.
Hemofili B.
Ved hemofili B mangler koagulasjonsfaktoren IX (antihemofil globulin B eller julefaktor). Også her er pasientene stort sett mannlige. Tidligere forekom hemofili B oftere i den engelske kongefamilien og i den russiske tsarfamilien. Derfor kalles det også "kongens sykdom".
Andre former for hemofili
I tillegg til hovedformene for hemofili A og B, er det andre arvelige koagulasjonsforstyrrelser. En av dem er von Willebrand-Jürgens syndrom, også kalt von Willebrand syndrom (vWS). Fokuset her er på von Willebrand -faktoren: denne koagulasjonsfaktoren er enten for liten eller defekt. Som med hemofili A og B, fører dette til en økt tendens til blødning (hemoragisk diatese). I Tyskland finnes von Willebrand-Jürgens syndrom hos opptil en prosent av befolkningen. I motsetning til de to hovedformene for hemofili påvirker denne koagulasjonsforstyrrelsen menn og kvinner likt.
I sjeldne tilfeller forekommer også andre koagulasjonsforstyrrelser, som er basert på mangel på koagulasjonsfaktorer. De har også en økt tendens til blødning (hemoragisk diatese). Disse inkluderer:
- Hemofili C: Mangel på funksjonell faktor XI
- Parahemophilia: mangel på fungerende faktor V
- Hypoprokonvertinemi: Mangel på funksjonell faktor VII
- Stuart Prower Factor Deficiency: Mangel på arbeidsfaktor X
- Hageman syndrom: mangel på funksjonsfaktor XII
- Fibrinasemangel: mangel på funksjonell faktor XIII
Hemofili: symptomer
Symptomene på hemofili A (faktor VIII -mangel) og hemofili B (faktor IX -mangel) er de samme - selv om forskjellige koagulasjonsfaktorer påvirkes i hvert enkelt tilfelle. Generelt kan man si: jo færre fungerende koagulasjonsfaktorer det er, jo mer uttalt er det kliniske bildet.
Hemofili: alvorlighetsgrader
Alvorlighetsgraden av hemofili avhenger av hvor mye aktiviteten til koagulasjonsfaktorene reduseres i forhold til deres funksjon hos friske mennesker. Hvis faktoraktiviteten bare er noe redusert, har de som er berørt vanligvis ingen symptomer. På den annen side, hos symptomatiske hemofiler, er faktoraktiviteten mer redusert, noe som forårsaker mer uttalte tegn. Det er tre alvorlighetsgrader av hemofili (A og B):
Mild hemofili:Faktoraktiviteten er 6 til 45 prosent av normal aktivitet hos en frisk person; rundt 20 prosent av alle mennesker med hemofili har denne alvorlighetsgraden. De som rammes merker ofte ikke mye av sykdommen sin. Det er derfor hemofili bare oppdages hos mange mennesker i ungdomsårene eller i voksen alder, når operasjoner eller større skader blør lenger enn forventet.
Moderat hemofili: Faktoraktivitet er 1 til 5 prosent av normal aktivitet; rundt 20 prosent av alle hemofile er også berørt. Symptomene blir vanligvis tydelige de første årene av livet med uvanlig lange blødninger og hyppige blåmerker. Som med mild hemofili, er blødning vanligvis et resultat av skader eller operasjoner. Spontan blødning er sjelden.
Alvorlig hemofili: Faktoraktivitet er mindre enn 1 prosent av normal aktivitet. Dette gjelder omtrent 55 prosent av alle hemofili. Hemofili er vanligvis allerede merkbar ved fødselen: fjerning av navlestrengen utløser massiv blødning. Kraftig neseblod er ikke uvanlig i barndommen. I tillegg kan selv de minste skadene eller støtene forårsake kraftig blødning under huden (blåmerker). Intern blødning er også mer vanlig, for eksempel smertefull blødning i de store leddene (som knær, albuer). Vanligvis har mange blødninger ingen åpenbar årsak (spontan blødning).
Hemofili: risiko og farer
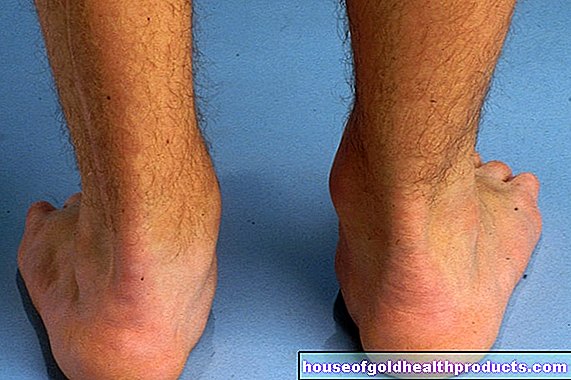
Blødning i leddene (hemartrose) forekommer ofte gjentatte ganger, spesielt ved alvorlig hemofili. Som et resultat kan det berørte leddet deformeres, slites ut for tidlig (slitasjegikt) og gradvis stivne. Personer med avansert hemartose kan knapt bevege armer og ben og er avhengige av rullestol.
En annen fare med hemofili er blødning i musklene: det kan skade muskelvevet og føre til muskelsvakhet.
I prinsippet kan indre blødninger ved hemofili forekomme i ethvert organområde. Blødning i hjernen er sjelden, men farlig: for eksempel kan det svekke evnen til å tenke og svekke konsentrasjonen. Alvorlig hjerneblødning kan til og med være dødelig! Blødning i magen kan også være livstruende under visse omstendigheter. Kraftig blødning i orofarynx kan påvirke luftveiene.
I tillegg til blødningstendenser (hemoragisk diatese), kan hemofili av en hvilken som helst alvorlighetsgrad også føre til sårhelingsforstyrrelser.
Von Willebrand-Juergens syndrom: Symptomer
Personer med von Willebrand-Jürgens syndrom har også en økt tendens til å blø. Mesteparten av tiden er det lett blødning under huden (blåmerker), blødende tannkjøtt eller langvarig blødning etter operasjon, for eksempel etter tannuttrekking. Men det er også pasienter som lider av kraftige blødninger.
Hemofili: behandling
Hemofili -behandling avhenger av hemofiliens type og alvorlighetsgrad. Den behandlende legen vil lage en passende terapiplan for hver pasient. I tillegg til medikamentell behandling kan han også anbefale generelle tiltak. For eksempel kan det være lurt, spesielt ved alvorlig hemofili, å ta fysisk omsorg og unngå visse (skadelige) idretter.
Hemofili A og B
I dag er såkalte faktor-konsentrater tilgjengelig for behandling av hemofili. Dette er konsentrater av blodkoagulasjonsfaktorer VIII (for hemofili A) eller IX (for hemofili B) hentet fra blodplasma eller genetisk konstruert. De må injiseres i en vene (intravenøst). Mange som lider, lærer å injisere faktorkonsentratet selv. Dette gir dem mye uavhengighet i behandlingen av sykdommen.
På 1960- og 1970 -tallet fikk mange mennesker i Tyskland hepatitt og / eller HIV fra forurensede faktorpreparater. Dette kan praktisk talt ikke skje i dag: blodplasma kontrolleres strengt og forbehandles før det administreres. Og med genetisk konstruerte faktor -konsentrater er det generelt ingen risiko for infeksjon.
Ved mild og moderat hemofili er administrering av faktorkonsentrat bare nødvendig om nødvendig (behandling på forespørsel): Antikoagulanten administreres for eksempel ved kraftigere blødninger eller før en planlagt operasjon. Mindre skader som slitasje trenger ikke å behandles med faktorkonsentrat. Blødningen kan vanligvis stoppes ved å trykke lett på blødningsområdet.
Personer med alvorlig hemofili må derimot injisere seg med faktorkonsentrat regelmessig (langtidsbehandling). Faktor VIII ved hemofili A administreres to til tre ganger i uken. Faktor IX ved hemofili B trenger vanligvis bare å injiseres en eller to ganger i uken på grunn av lengre oppbevaringstid i blodet.
Operasjoner og akutte skader
Før operasjoner (selv om en tannekstraksjon er planlagt), må forberedende behandling utføres for alle hemofili -pasienter. Et faktor -konsentrat administreres vanligvis for dette formålet. Dette er den eneste måten å forhindre alvorlige komplikasjoner fra overdreven blodtap under operasjonen.
Ved mild hemofili A, i stedet for faktorkonsentrater, kan et legemiddel som stabiliserer blodpropp gis før en planlagt operasjon. Dette inkluderer for eksempel desmopressin. Dette er et kunstig produsert protein. Det stimulerer frigjøring av lagret faktor VIII fra blodkar. Legemidlet kan bare brukes i noen få dager, ellers er butikkene snart tomme.
Tips: Før en planlagt prosedyre bør personer med hemofili diskutere med hematologen eller hemofilisenteret om hvilken forebyggende behandling som er tilrådelig i deres tilfelle.
Ved akutte skader i en nødssituasjon, i tillegg til lokale hemostasetiltak (f.eks. Trykkbandasjer), er administrering av faktorkonsentrat også nødvendig.
Faktorkonsentrat: komplikasjoner
Noen mennesker danner antistoffer (hemmere) mot koagulasjonsfaktorene i faktorkonsentratet. Denne såkalte hemmeren hemofili er betydelig mer vanlig hos personer med hemofili A enn hos personer med hemofili B. Inhibitorene inaktiverer den ekstra koagulasjonsfaktoren. Behandlingen er da ikke så effektiv som ønsket. Hemofili B truer også med alvorlige allergiske reaksjoner og andre komplikasjoner.
Mengden hemmere i blodet er gitt i den såkalte Bethesda-enheten (BE). Jo høyere BE -verdi, desto flere hemmere er det i pasientens blod.
Ved hemofili A kan en liten opphopning av hemmere ofte kompenseres ved å øke dosen av faktorkonsentrat. Hvis det dannes hemmere i store mengder, anbefales immuntoleransebehandling: pasienten får svært høye doser av den manglende koagulasjonsfaktoren i et komplekst terapiregime. Immunsystemet bør gradvis venne seg til dets tilstedeværelse og stoppe dannelsen av hemmere.
Den sjeldne hemmeren hemofili i hemofili B behandles annerledes. For eksempel mottar de berørte legemidler som påvirker immunsystemet (immunmodulatorer).
Smertestillende
Alvorlig hemofili kan forårsake alvorlig smerte for de berørte. For eksempel kan blødning i leddene være veldig smertefullt. Da hjelper smertestillende midler som ibuprofen. I kontrast er smertestillende acetylsalisylsyre (ASA) uegnet for hemofili: det øker tendensen til å blø enda mer (bivirkning av ASA).
von Willebrand-Juergens syndrom (VWS)
Ved von Willebrand-Jürgens syndrom skilles det mellom forskjellige typer, som behandles ulikt: I den såkalte type 1 gis den aktive ingrediensen desmopressin når det er nødvendig (før en operasjon eller ved akutt blødning). Det stimulerer frigjøring av lagrede koagulasjonsfaktorer.
Desmopressin brukes også i type 2; stoffet virker imidlertid ikke alltid her. Deretter foreskriver legen et faktorkonsentrat (med von Willebrand -faktor) i stedet.
Pasienter med VWS type 3 behandles alltid med et faktorkonsentrat.
Hemofili: årsaker og risikofaktorer
Hemofili er en medfødt genetisk lidelse som vanligvis er arvet. Det skjer sjeldnere spontant (som et resultat av en spontan genendring = spontan mutasjon).
Hos hemofili er den genetiske informasjonen som kreves for å produsere en funksjonell koagulasjonsfaktor feil: i hemofili A er det koagulasjonsfaktor VIII, i hemofili B, faktor IX. Resultatet av feil byggeplan er at den aktuelle koagulasjonsfaktoren ikke kan produseres i tilstrekkelig funksjonell mengde. Dette forstyrrer blodpropp: sår lukker seg ikke så raskt, så blødning tar uvanlig lang tid. Noen pasienter har også spontan blødning (uten noen åpenbar årsak).
Hemofili A og B: arv
Blåkopiene (genene) for alle deler av kroppen er på kromosomene. I kjernen i hver celle i kroppen er det 46 kromosomer, inkludert to kjønnskromosomer, som blant annet bestemmer kjønn. Kvinner har to X -kjønnskromosomer (XX): Ett X -kromosom arvet fra moren og ett fra faren. Menn, derimot, har et Y -kromosom arvet fra faren og et X -kromosom arvet fra moren (XY).
Genene for koagulasjonsfaktorene er på X -kromosomet. Hos kvinner som har to X -kromosomer, hvis en av dem inneholder en defekt plan for en koagulasjonsfaktor, kan dette vanligvis kompenseres for det andre X -kromosomet. De er derfor stort sett symptomfrie gjennom hele livet.
Hvis en slik kvinne får et barn, sender hun det ene av de to X -kromosomene videre til avkommet. Sannsynligheten for at dette er kopien med den defekte planen er 50 prosent. Ved å videreformidle den genetiske defekten, blir mor bæreren (bæreren) av hemofili. Hvis det er en sønn, vil han bli født som en hemofili. En datter, derimot, blir vanligvis en potensiell bærer etter tur.
I sjeldne tilfeller dannes ikke den relevante koagulasjonsfaktoren tilstrekkelig selv i hunnledere. Sår etter skader eller operasjoner kan da blø i lang tid. Det er enda mer sjelden at en jente arver et defekt X -kromosom fra begge foreldrene. Dette er mest i forbindelse med den arvelige sykdommen Turners syndrom. De berørte jentene viser hele bildet av hemofili.
Von Willebrand-Juergens syndrom
I von Willebrand-Jürgens syndrom viser instruksjonene for von Willebrand-faktoren (vWF) en mutasjon: koagulasjonsfaktoren er for liten eller defekt. Genmutasjonen kan forekomme hos både menn og kvinner.
Hemofili: undersøkelser og diagnose
Hvis noen ofte har spontan blødning (som neseblod) eller blåmerker, kan dette være en indikasjon på hemofili. Denne mistanken er spesielt åpenbar hvis familien allerede har kjent tilfeller av hemofili. De berørte bør få den økte blødningstendensen avklart av en lege. Det første kontaktpunktet hvis du mistenker at hemofili er fastlegen din:
Legen vil først ta pasientens medisinske historie (anamnese) i samtale med pasienten: Han har symptomene beskrevet i detalj, spør om eventuelle underliggende sykdommer og om det er kjente tilfeller av hemofili i familien.
Laboratorietester er spesielt viktige for å avklare en mulig hemofili. Legen tar en blodprøve fra pasienten for å få den undersøkt på laboratoriet for forskjellige parametere: Ved hemofili er den såkalte aPTT (aktivert delvis tromboplastintid) lengre enn hos friske mennesker. I kontrast er den såkalte Quick-verdien (tromboplastintid, TPZ) og plasmatrombintiden (PTZ) generelt normal; de er bare forlenget ved alvorlig hemofili. Antall blodplater (trombocytter) og den såkalte blødningstiden er også normal ved hemofili A og B. I von Willebrand-Jürgens syndrom derimot, forlenges blødningstiden. Blødningstid er tiden det tar før blødningen stopper.
For å kunne bestemme utvetydig hemofili A eller B, må koagulasjonsfaktorers aktivitet (VIII, IX) analyseres. En spesialist i hematologi eller et spesialisert medisinsk senter (hemofilisenter) er ansvarlig for dette.
Hemofili: test hos nyfødte og ufødte babyer
Hvis en familie allerede har utviklet hemofili, blir koagulering av mannlige nyfødte vanligvis sjekket umiddelbart etter fødselen. På denne måten kan hemofili oppdages på et tidlig stadium. Du kan også se etter hemofili under graviditet.
Hvis en kvinne mistenker at hun har en genetisk disposisjon for hemofili og derfor er en potensiell bærer, kan dette avklares med en genetisk test.
Hemofili: sykdomsforløp og prognose
Hemofili er ennå ikke kurert. Pasienter må håndtere mangelen på koagulasjonsfaktorer gjennom livet. Ved hjelp av faktorkonsentrater kan de imidlertid vanligvis leve et stort sett normalt liv.
Hvis den ikke behandles, fører moderat og alvorlig hemofili ofte til alvorlige komplikasjoner. Blødning i musklene kan for eksempel forårsake muskelskade. Blødning i leddene kan føre til slitasjegikt og leddstivning. Slike komplikasjoner kan unngås hvis hemofili oppdages tidlig og behandles konsekvent.
Tags.: narkotika hud palliativ medisin